diff --git a/.github/CONTRIBUTING.md b/.github/CONTRIBUTING.md
new file mode 100644
index 00000000..90a7699e
--- /dev/null
+++ b/.github/CONTRIBUTING.md
@@ -0,0 +1,68 @@
+OpenMS Documentation Contributing Guidelines
+============================================
+
+Hi there!
+
+Thank you for thinking about enhancing OpenMS Documentation further.
+
+Please feel free to:
+
+1. Create a bug report or feature request in OpenMS Documentation, [here](https://github.com/OpenMS/OpenMS-docs/issues).
+2. Create a pull request in [OpenMS Documentation](https://github.com/OpenMS/OpenMS-docs) with the change you're proposing.
+
+For any questions, drop us a ping in [OpenMS Gitter](https://gitter.im/OpenMS/OpenMS).
+
+## Create a Pull Request(PR)
+
+1. Fork this repository.
+2. Add the change in your fork.
+3. Create a pull request to [OpenMS/OpenMS-Docs](https://github.com/OpenMS/OpenMS-docs/tree/staging) `staging` branch.
+4. Make sure you attach a few screenshots as to how your change looks like.
+5. If this change belongs to [OpenMS API reference](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html),
+ please create a pull request [here](https://github.com/OpenMS/OpenMS/tree/develop/doc).
+
+## Documentation content writing conventions
+
+1. Please prefer active voice.
+2. Restrict the line length to 120 characters, including space(s).
+3. Title and H1 heading should be in `Title Case`. Follow `Sentence case`, until otherwise stated.
+4. Write as OpenMS is writing for itself as an object and subject.
+5. Use American English.
+6. Use `backtick`(s) for formatting code, library name, files, and path.
+7. Use **bold** for product name, object name, an independent entity.
+8. Use **bold** for menu title in an application.
+9. Link to glossary terms using {term}\`this is a glossary term\`.
+10. OpenMS documentation uses following Admonitions
+ - Hint
+ - Important
+ - Note
+ - Warning
+ - Tip
+ - See Also
+
+ Example of these are present in documentation, please follow them.
+11. Always specify lexers for code blocks.
+12. Format keyboard strokes using `qwerty-keyboard-button`, as an example, please see [this](../docs/tutorials/TOPP/hotkeys-table.md) file.
+13. Be nice, polite, and respectful.
+
+### Naming files
+
+1. The title of the page should be the `name-of-the-file.md`.
+2. Prefer writing in markdown, with an `.md` file extension; both reStructureText and markdown is supported in OpenMS Documentation.
+
+### Images and figures
+
+For images and figures:
+
+1. Add a screenshot of the window.
+2. In tutorial, align images in center. Other instructions, should have alighment to left.
+3. Please set the size to `500px` of images added in step-by-step guides or instructions.
+
+## OpenMS documentation contributors
+
+Thank you for your contribution!
+
+Finally, please add your name below:
+
+1. OpenMS Team
+
diff --git a/.github/ISSUE_TEMPLATE/bug-report.md b/.github/ISSUE_TEMPLATE/bug-report.md
new file mode 100644
index 00000000..4d28f34d
--- /dev/null
+++ b/.github/ISSUE_TEMPLATE/bug-report.md
@@ -0,0 +1,45 @@
+---
+name: Bug report
+about: Report something that is broken or incorrect in documentation
+labels: bug
+---
+
+
+
+## Read Existing Documentation
+
+I have checked the following places for the error:
+
+- [ ] [OpenMS Documentation Troubleshooting]()
+- [ ] [OpenMS API Developer FAQs]()
+
+## Create OpenMS Bug Report
+
+I have created the bug report in OpenMS for any pipeline errors.
+
+- [ ] [OpenMS Open Issues](https://github.com/OpenMS/OpenMS/issues)
+
+## Description of the documentation bug
+
+- [ ] I have attached screenshots of the incorrect documentation
+
+
+
+## Expected behaviour
+
+
+
+## OpenMS Installation
+
+- Version:
+
+## Additional context
+
+
diff --git a/.github/ISSUE_TEMPLATE/feature-request.md b/.github/ISSUE_TEMPLATE/feature-request.md
new file mode 100644
index 00000000..ebdb28fd
--- /dev/null
+++ b/.github/ISSUE_TEMPLATE/feature-request.md
@@ -0,0 +1,35 @@
+---
+name: Feature request
+about: Suggest an idea for the OpenMS Documentation
+labels: enhancement
+---
+
+
+
+## Is your feature request related to a OpenMS API Reference Documentation? Please describe.
+
+
+- [ ] I have attached screenshot describing the problem, if applicable.
+
+## Describe the documentation enhancement you'd like
+
+
+
+## Describe alternatives you've considered
+
+
+
+## Additional context
+
+
diff --git a/.github/PULL_REQUEST_TEMPLATE.md b/.github/PULL_REQUEST_TEMPLATE.md
new file mode 100644
index 00000000..f29a1de1
--- /dev/null
+++ b/.github/PULL_REQUEST_TEMPLATE.md
@@ -0,0 +1,23 @@
+
+
+## Describe the change
+
+
+
+## PR checklist
+
+- [ ] I have added description of the change I'm proposing in the OpenMS Documentation.
+- [ ] I have read and followed [OpenMS Documentation Contributing guidelines](CONTRIBUTING.md).
+- [ ] I have attached a screenshot of the relevant area after this change.
+- [ ] `CHANGELOG.md` is updated.
+- [ ] I have added my name in [CONTRIBUTING.md](CONTRIBUTING.md#openms-documentation-contributors).
diff --git a/.readthedocs.yaml b/.readthedocs.yaml
new file mode 100644
index 00000000..1f2cc099
--- /dev/null
+++ b/.readthedocs.yaml
@@ -0,0 +1,17 @@
+version: 2
+
+# Build from the docs/ directory with Sphinx
+sphinx:
+ configuration: conf.py
+
+# Explicitly set the version of Python and its requirements
+version: 2
+
+build:
+ os: ubuntu-20.04
+ tools:
+ python: "3.9"
+
+python:
+ install:
+ - requirements: requirements.txt
diff --git a/CHANGELOG.md b/CHANGELOG.md
new file mode 100644
index 00000000..1465d6fd
--- /dev/null
+++ b/CHANGELOG.md
@@ -0,0 +1,8 @@
+OpenMS 2.8.0 Documentation which includes
+
+1. OpenMS Introductory Pages
+2. Installation Instructions
+3. TOPP and TOPPAS tutorials
+4. TOPP Introductory Documentation
+5. User QuickStart Guide
+6. Contacting OpenMS
diff --git a/Makefile b/Makefile
new file mode 100644
index 00000000..de72f3b7
--- /dev/null
+++ b/Makefile
@@ -0,0 +1,21 @@
+# Minimal makefile for Sphinx documentation
+#
+
+# You can set these variables from the command line.
+SPHINXOPTS =
+SPHINXBUILD = sphinx-build
+SPHINXPROJ = openms
+SOURCEDIR = docs
+BUILDDIR = build
+
+# Put it first so that "make" without argument is like "make help".
+help:
+ @$(SPHINXBUILD) -M help "$(SOURCEDIR)" "$(BUILDDIR)" $(SPHINXOPTS) $(O)
+
+
+.PHONY: help Makefile
+
+# Catch-all target: route all unknown targets to Sphinx using the new
+# "make mode" option. $(O) is meant as a shortcut for $(SPHINXOPTS).
+%: Makefile
+ @$(SPHINXBUILD) -M $@ "$(SOURCEDIR)" "$(BUILDDIR)" $(SPHINXOPTS) $(O)
diff --git a/README.md b/README.md
index 0d2cf8c9..22f99c81 100644
--- a/README.md
+++ b/README.md
@@ -1 +1,7 @@
-# OpenMS-docs
\ No newline at end of file
+OpenMS Documentation
+====================
+
+You're reading OpenMS `staging` documentation! If you think, you're at a wrong place, go to [OpenMS Documentation](https://staging-openms.readthedocs.io/en/staging/docs/index.html).
+
+Please read our [contributing guidelines](.github/CONTRIBUTING.md), before starting with
+contributing to OpenMS Documentation.
diff --git a/_static/css/custom.css b/_static/css/custom.css
new file mode 100644
index 00000000..50ef9f6b
--- /dev/null
+++ b/_static/css/custom.css
@@ -0,0 +1,3 @@
+div.highlight {
+ background-color: #f6f8fa;
+}
diff --git a/assets/logo/OpenMS.svg b/assets/logo/OpenMS.svg
new file mode 100644
index 00000000..0a25fbaa
--- /dev/null
+++ b/assets/logo/OpenMS.svg
@@ -0,0 +1,333 @@
+
+
\ No newline at end of file
diff --git a/assets/logo/OpenMS_transparent_background.png b/assets/logo/OpenMS_transparent_background.png
new file mode 100644
index 00000000..bf1f774a
Binary files /dev/null and b/assets/logo/OpenMS_transparent_background.png differ
diff --git a/conf.py b/conf.py
new file mode 100644
index 00000000..d69c157b
--- /dev/null
+++ b/conf.py
@@ -0,0 +1,107 @@
+# Configuration file for the Sphinx documentation builder.
+#
+# This file only contains a selection of the most common options. For a full
+# list see the documentation:
+# https://www.sphinx-doc.org/en/master/usage/configuration.html
+
+# -- Path setup --------------------------------------------------------------
+
+# If extensions (or modules to document with autodoc) are in another directory,
+# add these directories to sys.path here. If the directory is relative to the
+# documentation root, use os.path.abspath to make it absolute, like shown here.
+#
+# import os
+# import sys
+# sys.path.insert(0, os.path.abspath('.'))
+
+
+# -- Project information -----------------------------------------------------
+
+project = 'OpenMS'
+copyright = '2022, OpenMS Team'
+author = 'OpenMS Team'
+
+# -- General configuration ---------------------------------------------------
+
+# Add any Sphinx extension module names here, as strings. They can be
+# extensions coming with Sphinx (named 'sphinx.ext.*') or your custom
+# ones.
+extensions = [
+ 'sphinx_copybutton',
+ 'sphinx.ext.autodoc',
+ 'sphinx.ext.autosummary',
+ 'sphinx.ext.autosectionlabel',
+ 'myst_parser',
+ 'notfound.extension',
+ 'sphinxcontrib.images',
+ 'sphinx_inline_tabs',
+]
+
+myst_enable_extensions = [
+ "tasklist",
+ "dollarmath",
+ "amsmath",
+ "colon_fence",
+ "linkify",
+ "replacements",
+ "linkify_fuzzy_links",
+]
+
+# Generate header anchors for cross-linking in markdown until depth n
+myst_heading_anchors = 3
+
+autosummary_generate = True
+autosummary_imported_members = True
+
+# Add any paths that contain templates here, relative to this directory.
+templates_path = ['_templates']
+
+# The suffix(es) of source filenames.
+# You can specify multiple suffix as a list of string:
+#
+source_suffix = ['.rst', '.md']
+
+# List of patterns, relative to source directory, that match files and
+# directories to ignore when looking for source files.
+# This pattern also affects html_static_path and html_extra_path.
+exclude_patterns = ['_build', 'Thumbs.db', '.DS_Store']
+
+# The version info for the project you're documenting, acts as replacement for
+# |version| and |release|, also used in various other places throughout the
+# built documents.
+#
+# The short X.Y version.
+version = '2.8.0'
+# The full version, including alpha/beta/rc tags.
+release = '2.8.0'
+
+# -- Options for HTML output -------------------------------------------------
+
+# The theme to use for HTML and HTML Help pages. See the documentation for
+# a list of builtin themes.
+#
+html_theme = 'furo'
+html_logo = 'assets/logo/OpenMS_transparent_background.png'
+html_theme_options = {
+ "navigation_with_keys": True,
+ "light_css_variables": {
+ "font-stack--monospace": "Consolas, monospace",
+ "font-size--small": "90%",
+ "toc-font-size": "87.5%"
+ },
+}
+pygments_style = 'sas'
+
+pygments_dark_style = 'rrt'
+
+
+# Add any paths that contain custom static files (such as style sheets) here,
+# relative to this directory. They are copied after the builtin static files,
+# so a file named "default.css" will overwrite the builtin "default.css".
+html_static_path = ['_static']
+
+html_css_files = [
+ 'css/custom.css',
+]
+
+root_doc = 'docs/index'
diff --git a/docs/additional-resources/developer-guidelines-for-addding-new-dependent-libraries.md b/docs/additional-resources/developer-guidelines-for-addding-new-dependent-libraries.md
new file mode 100644
index 00000000..47707d6e
--- /dev/null
+++ b/docs/additional-resources/developer-guidelines-for-addding-new-dependent-libraries.md
@@ -0,0 +1,68 @@
+Developer Guidelines For Adding New Dependent Libraries
+======================================================
+
+## Our dependency library philosophy
+
+In short, requirements for adding a new library are:
+- indispensable functionality
+- license compatibility
+- availability for all platforms
+
+## Indispensable functionality
+
+In general, adding a new dependency library (of which we currently have more than a handful, e.g. Xerces-C or ZLib)
+imposes a significant integration and maintenance effort. Thus, the new library should add **indispensable functionality**.
+If the added value does not compensate for the overhead, alternative solutions encompass:
+
+- write it yourself and add to the OpenMS library (i.e. its repository) directly
+- write a TOPPAdapter which calls an external executable (placing the burden on the user to supply the executable)
+
+## License compatibility
+
+OpenMS has a BSD-3 clause license and we try hard to remove dependencies of GSL-like libraries. Therefore, a new library
+with e.g. LGPL-2 would be prohibitive.
+
+
+## C++ standard compatibility
+
+New dependency libraries needs to be compatible and therefore compilable with the same C++ standard as OpenMS.
+
+## Availability for all platforms
+
+OpenMS has been designed for Windows, macOS, and Linux. Therefore, the new dependency library needs to be designed for
+these platforms.
+
+- on **WindowsOS** this usually means, adding the new library to the Contrib in debug and release variants. In short all
+ recent versions of Visual Studio (VS2008 and onwards) must be supported (or support must be added). This encompasses
+ - a solution file (which can be either statically present or generated by a meta-system like CMake) is available
+ - The library actually compiles and is linked to the **dynamic** VS-C++ runtime lib (since this is what the OpenMS lib will
+ link to as well - combining static and dynamic links will lead to linker errors or segfaults).
+
+- on **macOS** it should be ensured that the library can be build on recent macOS versions (> 10.10) compiled using the
+ mac specific _libc++_. Ideally the package should be available via **HomeBrew** or **MacPorts** so we can directly use
+ those libraries instead of shipping them via the contrib. Additionally, the MacPorts and HomeBrew formulas for building
+ the libraries can serve as blueprints on how to compile the library in a generic setting inside the contrib which should
+ also be present.
+
+- on **Linux** since we (among other distributions) feature an OpenMS Debian package which requires that all dependencies
+ of OpenMS are available as Debian package as well, the new library must be available (or made available) as Debian
+ package or linked statically during the OpenMS packaging build.
+
+## How to add it to the contrib build
+
+Add a CMake file to `OpenMS/contrib` into the `libraries.cmake` folder on how to build the library. Preferably of course
+the library supports building with CMake (see Xerces) which makes the script really easy. It should support static and
+dynamic builds on every platform. Add the compile flag for position independent code (e.g. `-fpic`) in the static version.
+Add patches in the *patches* folder and call them with the macros in the `macros.cmake` file. Create patches with
+`diff -Naur original_file my_file > patch.txt`. If there are problems during applying a patch, make sure to double check
+filepaths in the head of the patch and the call of the patching macro in CMake.
+
+- All the libraries need to go into (e.g. copied/installed/moved) to `$buildfolder/lib`
+- All the headers under `$buildfolder/include/$libraryname` (the only exception to leave out the library name subfolder
+ is when the `Find$libraryname.cmake` does not support this subfolder e.g. because system libraries are not structured
+ like this, see boost).
+- All needed files into `$buildfolder/share/$libraryname`
+
+Then test the build on your platform including a subsequent build of OpenMS using that library. Submit a pull request to
+`OpenMS/contrib`. Submit a pull request to `OpenMS/OpenMS` that updates the contrib submodule. Make sure the libraries
+are correctly shipped in pyOpenMS and the packages (especially dynamic libraries and especially on Windows).
diff --git a/docs/additional-resources/external-code-using-openms.md b/docs/additional-resources/external-code-using-openms.md
new file mode 100644
index 00000000..556c262d
--- /dev/null
+++ b/docs/additional-resources/external-code-using-openms.md
@@ -0,0 +1,120 @@
+External Code using OpenMS
+==========================
+
+If OpenMS' TOPP and UTILS tools are not enough in a certain scenario, you can either request a change to OpenMS, if you
+feel this functionality is useful for others as well, or modify/extend OpenMS privately. For the latter, there are multiple
+ways to do this:
+
+- Modify the developer version of OpenMS by changing existing tools or adding new ones.
+- Use an **External Project** to write a new tool, while not touching OpenMS itself (see below on how to do that).
+
+Once you've finished your new tool, and it only needs to run on the development machine. To ship it to a new client machine,
+see, read further in this document.
+
+## Compiling external code
+
+It is very easy to set up an environment to write your own programs using OpenMS. Make sure to downloaded and installed
+the source package of OpenMS/TOPP properly.
+
+```{note}
+You cannot use the `install` target when working with the development version of OpenMS, it must be built and used within
+the build tree.
+```
+
+All important compiler settings and preprocessor definitions along with the OpenMS library are available. The most
+important variables are:
+
+- `OpenMS_INCLUDE_DIRECTORIES`: all include directories containing OpenMS headers
+- `OPENMS_ADDCXX_FLAGS`: preprocessor macros we require written as `(-DMACRO1 -DMACRO2)`
+
+and the OpenMS target itself (which you can link against).
+
+The example that follows will be explained in details:
+
+```cpp
+### example CMakeLists.txt to develop C++ programs using OpenMS
+project("Example_Project_using_OpenMS")
+cmake_minimum_required(VERSION 3.0)
+
+## list all your executables here (a corresponding .cpp file should exist, e.g. Main.cpp)
+set(my_executables
+ Main
+)
+
+## list all classes here, which are required by your executables
+## (all these classes will be linked into a library)
+set(my_sources
+ ExampleLibraryFile.cpp
+)
+
+## find OpenMS configuration and register target "OpenMS" (our library)
+find_package(OpenMS)
+## if the above fails you can try calling cmake with -D OpenMS_DIR=/path/to/OpenMS/
+## or modify the find_package() call accordingly
+## find_package(OpenMS PATHS "
+cmake -G "" .
+```
+
+For more information visit the website of cmake at cmake.org and consult the documentation.
+
+```{important}
+Have fun coding with OpenMS!
+```
+
+## Shipping external code to a new machine
+
+If you've modified OpenMS itself and not used an external project use our installer scripts, to build your own OpenMS
+installer for your platform (see our internal FAQ which is built using "make doc_internal") and ship that to a client
+machine.
+
+If you've used an external project and have a new executable (+ an optional new library), use the installer approach as
+well, and manually copy the new executable to the `TOPP/UTILS` binary directory (e.g. on Windows this could be
+`c:/program files/OpenMS/bin`, on Linux it could be `/bin`.
+
+If you do not use the installer, copy all required files manually, plus a few extra steps, see below. What needs to be
+done is a little platform dependent, thus very cumbersome to explain. Look at the cmake installer scripts, to see whats
+required (for macOS and Linux see `OpenMS/cmake/package*.cmake`).
+
+In short:
+
+- copy the `OpenMS/share/OpenMS` directory to the client machine (e.g `/share`) and set the environment
+ variable `OPENMS_DATA_PATH` to this directory
+- copy the OpenMS library (`OpenMS.dll` for Windows or `OpenMS.so/.dylib` for Linux/macOS) to `/bin`.
+- copy all Qt4 libraries to the client `/bin` or on Linux/macOS make sure you have installed the Qt4 package
+- [Windows only] copy Xerces dll (see `contrib/lib`) to `/bin`
+- [Windows only] install the VS redistributable package (see Microsoft Homepage) on the client machine which corresponds
+ to the VS version that was used to compile your code (use the correct redistributable package!, i.e., architecture
+ 32|64bit, VS version, VS Service Pack version). If you choose the wrong redistributable package, you will get
+ "Application failed to initialize properly..." error messages.
diff --git a/docs/additional-resources/openms-git-workflow.md b/docs/additional-resources/openms-git-workflow.md
new file mode 100644
index 00000000..81bdf1a6
--- /dev/null
+++ b/docs/additional-resources/openms-git-workflow.md
@@ -0,0 +1,200 @@
+OpenMS Git Workflow
+===================
+
+Before getting started, install latest version of git to avoid problems like GitHub https authentication errors
+(see [Troubleshooting cloning errors](https://docs.github.com/en/repositories/creating-and-managing-repositories/troubleshooting-cloning-errors) and a solution using [ssh](https://docs.github.com/en/get-started/getting-started-with-git/about-remote-repositories)).
+
+OpenMS follows the [git flow workflow](https://nvie.com/posts/a-successful-git-branching-model/). The difference is that
+merge commits are managed via pull requests instead of creating merge commits locally.
+
+## Naming conventions
+
+Naming conventions for the following apply:
+
+* A **local repository** is the repository that lies on your hard drive after cloning.
+* A **remote repository** is a repository on a git server such as GitHub.
+* A **fork** is a copy of a repository. Forking a repository allows you to freely experiment with changes without affecting the original project.
+* **Origin** refers to a remote repository that you have forked. Call this repository `https://github.com/_YOURUSERNAME_/OpenMS`.
+* **Upstream** refers to the original remote OpenMS repository. Call this repository `https://github.com/OpenMS/OpenMS`.
+
+## Create fork
+
+Start by [forking](https://docs.github.com/en/get-started/quickstart/fork-a-repo) the OpenMS repository.
+
+To create a fork, click **Fork** under the main menu as shown below.
+
+
+
+## Clone your fork
+
+To obtain a local repository copy, clone your fork using:
+
+```bash
+$ git clone https://github.com/_YOURUSERNAME_/OpenMS.git
+```
+
+This will clone your fork (correctly labelled `origin` by default) into a local copy on your computer.
+
+```{note}
+To use `git clone git@github.com:_YOURUSERNAME_/OpenMS.git`, make sure you have [SSH key added to your GitHub account](https://docs.github.com/en/authentication/connecting-to-github-with-ssh/adding-a-new-ssh-key-to-your-github-account).
+```
+## Link remote branches to your local working repository
+
+After cloning your fork, your local repository should be named origin. Validate this by executing:
+
+```bash
+$ git remote -v
+ origin https://github.com/_YOURUSERNAME_/OpenMS.git (fetch)
+ origin https://github.com/_YOURUSERNAME_/OpenMS.git (push)
+```
+
+Sync data between your local copy, your fork (`origin`) and the remote original OpenMS/OpenMS repository (`upstream`)
+by using the following command:
+
+```bash
+$ git remote add upstream https://github.com/OpenMS/OpenMS.git
+```
+Verify that upstream was added correctly by calling:
+
+```bash
+$ git remote -v
+ origin https://github.com/_YOURUSERNAME_/OpenMS.git (fetch)
+ origin https://github.com/_YOURUSERNAME_/OpenMS.git (push)
+ upstream https://github.com/OpenMS/OpenMS.git (fetch)
+ upstream https://github.com/OpenMS/OpenMS.git (push)
+
+```
+
+Fetch changes and new branches from your fork (`origin`) as well as from the central, upstream OpenMS repository by executing:
+
+```bash
+$ git fetch upstream
+$ git fetch origin
+```
+or
+
+```bash
+$ git fetch --all
+```
+
+Create a local branch using the following:
+
+```bash
+$ git checkout -b
+```
+
+Call `git branch -va` to display the status of local and remote branches. You should see an output that looks like this:
+
+```bash
+$ git branch -va
+* develop 349ec48 Merge pull request #691 from cbielow/MGF_fix
+ feature/my_shiny_new_feature 3c05538 [FEATURE] added option to keep, ensure or reassign UIDs during conversion
+ remotes/origin/SILACAnalyzer 3ceae38 Fixed test.
+ remotes/origin/antilope 3fe5aa3 git-svn-id: https://open-ms.svn.sourceforge.net/svnroot/open-ms/branches/antilope@12117 6adb6e08-d915-0410-941f-83917bcadc18
+ remotes/origin/develop 349ec48 Merge pull request #691 from cbielow/MGF_fix
+ remotes/origin/master b182ba5 [NOP] first commit after SVN import to git
+ remotes/origin/msnovogen 93a5e4c [OPT] For faster access to specific amino acids a ResidueServer was added.
+ remotes/upstream/HEAD -> upstream/develop
+ remotes/upstream/SILACAnalyzer 3ceae38 Fixed test.
+ remotes/upstream/antilope 3fe5aa3 git-svn-id: https://open-ms.svn.sourceforge.net/svnroot/open-ms/branches/antilope@12117 6adb6e08-d915-0410-941f-83917bcadc18
+ remotes/upstream/develop 349ec48 Merge pull request #691 from cbielow/MGF_fix
+ remotes/upstream/master b182ba5 [NOP] first commit after SVN import to git
+ remotes/upstream/msnovogen 93a5e4c [OPT] For faster access to specific amino acids a ResidueServer was added.
+```
+
+## Keep your fork in sync
+
+Keep your fork (`origin`) in sync with the OpenMS repository (`upstream`) by following the [GitHub instructions](https://docs.github.com/en/pull-requests/collaborating-with-pull-requests/working-with-forks/syncing-a-fork).
+In summary, to keep your fork in sync:
+1. Fetch changes from upstream and update your local branch.
+2. Push your updated local branch to your fork (`origin`).
+
+```{tip}
+To keep track of others repositories, use `git fetch --all --prune` to update them as well. The option `--prune` tells
+git to automatically remove tracked branches if they got removed in the remote repository.
+```
+
+```bash
+$ git fetch --all --prune
+$ git checkout develop
+$ git merge --ff-only upstream/develop
+$ git push origin develop
+```
+Feel free to experiment within your fork. However, for your code needs to meet OpenMS quality standards to be merged
+into the OpenMS repository,
+
+Follow these rules:
+* Never commit directly to the `develop` or `master` branches as it will complicate the merge.
+* Try to start every feature from develop and not base features on other features.
+* Name the OpenMS remote `upstream` and always push directly to `origin` (`git push origin `).
+* When updating your fork, consider using `git fetch upstream` followed by `git merge --ff-only upstream/develop` to
+ avoid creating merge commits in `develop`.
+* If you never commit to `develop` this should always succeed and (if a commit accidentally went to develop) warn you
+ instead of creating a merge commit.
+
+## Create new feature
+
+All features start from `develop`.
+
+```bash
+$ git checkout develop
+$ git checkout -b feature/your-cool-new-feature
+```
+All commits related to this feature will then go into the branch `feature/your-cool-new-feature`.
+
+## Keeping your feature branch in sync with develop branch
+
+While working on your feature branch, it is usual that development continues and new features get integrated into the
+main development branch. This means your feature branch lags behind `develop`. To get your feature branch up-to-date,
+rebase your feature branch on `develop` using:
+
+```bash
+$ git checkout feature/myfeaturebranch
+$ git rebase develop
+```
+
+The above commands:
+
+1. Performs a rewind of your commits until the branching point.
+2. Applies all commits that have been integrated into `develop`.
+3. Reapplies your commits on top of the commits integrated into `develop`.
+
+For more information, refer to a [visual explanation of rebasing](http://git-scm.com/book/en/v2/Git-Branching-Rebasing).
+
+```{tip}
+Do not rebase published branches (e.g. branches that are part of a pull request). If you created a pull request, you
+should only add commits in your feature branch to fix things that have been discussed. After your pull request contains
+all fixes, you are ready to merge the pull request into develop without rebasing (see e.g. rebase-vs-merge).
+```
+
+## Adding a feature to OpenMS
+
+Features that should go into the main development line of OpenMS should be integrated via a [pull request](https://docs.github.com/en/pull-requests/collaborating-with-pull-requests/proposing-changes-to-your-work-with-pull-requests/about-pull-requests). This allows
+the development community of OpenMS to discuss the changes and suggest possible improvements.
+
+After opening the pull request via the GitHub web site, GitHub will try to create the pull request against the branch
+that you branched off from. Please check the branch that you are opening the pull request against before submitting the
+pull request. If any changes are made, a new pull request is required. Select
+**Allow others to make changes to this pull request** so that maintainers can directly help to solve problems.
+
+Open pull requests only after checking code-style, documentation and passing tests. Pull requests that do not pass CI
+or code review will not be merged until the problems are solved. It is recommended that you read the
+[pull request guidelines](pull-request-checklist.md) before you submit a pull request.
+
+## Update git submodules
+
+Start in your local `OpenMS/OpenMS` repository (on your feature/pull request branch).
+
+The following example uses a submodule called `THIRDPARTY`.
+
+```bash
+$ git submodule update --init THIRDPARTY
+$ cd THIRDPARTY
+# yes, in the submodules the default remote is origin
+# usually you want to pull the changes from master (e.g. after your pull request to OpenMS/THIRDPARTY has been merged)
+$ git pull origin master
+$ cd ..
+$ git status
+# Make sure that you see "modified: THIRDPARTY (new commits)"
+$ git commit -am "updated submodule"
+```
diff --git a/docs/additional-resources/pull-request-checklist.md b/docs/additional-resources/pull-request-checklist.md
new file mode 100644
index 00000000..ccb6487c
--- /dev/null
+++ b/docs/additional-resources/pull-request-checklist.md
@@ -0,0 +1,44 @@
+Pull Request Checklist
+======================
+
+Before opening a pull request, check the following:
+
+1. **Does the code build?**
+ Execute `make` (or your build system's equivalent, e.g., `cmake --build . --target ALL_BUILD --config Release` on Windows).
+2. **Do all tests pass?**
+ To check if all tests have passed, execute `ctest`.
+ If a test that is unrelated to your changes fails, check the [nightly builds](http://cdash.openms.de/index.php?project=OpenMS)
+ to see if the error is also in `develop`. If the error is in develop, [create a github issue](write-and-label-github-issues.md).
+3. **Is the code documented?**
+ Document all new classes, including their methods and parameters.
+ It is also recommended to document non-public members and methods.
+4. **Does the code introduce changes to the API?**
+ If the code introduces changes to the API, make sure that the documentation is up-to-date and that the Python bindings
+ (pyOpenMS) still work. For each change in the C++ API, make a change in the Python API wrapper via the `pyOpenMS/pxds/` files.
+5. **Have you completed regression testing?**
+ Make sure that you include a test in the test suite for:
+ - Public methods of a class
+ - TOPP tools
+ - Bug fixes
+
+Make sure to:
+
+- **Rebase before you open a pull request.**
+ To include all recent changes, rebase your branch on `develop` before opening a pull request.
+ If you pushed your branch to `origin` before rebasing, git will most likely tell you after the rebase that your
+ local branch and the remote branch have diverged. If you are sure that the remote branch does not contain any local
+ commits in the rebased version, you can safely push using `git push -f origin ` to enforce overwrite. If
+ not, contact your local git expert on how to get the changes into your local branch.
+
+- **Capture similar changes in a single commit**
+ Each commit should represent one logical unit. Consolidate multiple commits if they belong together or split single
+ commits if they are unrelated. For example, committing code formatting together with a one-line fix makes it very hard
+ to figure out what the fix was and which changes were inconsequential.
+
+* **Create a pull request for a single feature or bug**
+ If you have multiple features or fixes in a pull request, you might get asked to split your request and open multiple
+ pull requests instead.
+
+* **Describe what you have changed in your pull request.**
+ When opening the pull request, give a detailed overview of what has changed and why. Include a clear rationale for the
+ changes and add benchmark data if available. See [this request](https://github.com/bitly/dablooms/pull/19) for an example.
diff --git a/docs/additional-resources/reporting-bugs-and-issues.md b/docs/additional-resources/reporting-bugs-and-issues.md
new file mode 100644
index 00000000..3ac63aeb
--- /dev/null
+++ b/docs/additional-resources/reporting-bugs-and-issues.md
@@ -0,0 +1,19 @@
+Reporting Bugs and Issues
+=========================
+
+A list of known issues in the current OpenMS release can be found [here](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/known_dev_bugs.html). Please check if your OpenMS version matches the current version and if the bug
+has already been reported.
+
+In order to report a new bug, please create a [GitHub issue](write-and-label-github-issues.md) or [contact us](../contact-us.md).
+
+Include the following information in your bug report:
+
+1. The command line (i.e. call) including the TOPP tool and the arguments you used, or the steps you followed in a GUI
+ tool (e.g. TOPPView) - e.g. `FeatureFinderCentroided -in myfile.mzML -out myfile.featureXML`.
+2. The output of OpenMS/TOPP (or a screenshot in case of a GUI problem).
+3. Operating system (e.g. "Windows XP 32 bit", "Win 7 64 bit", "Fedora 8 32 bit", "macOS 10.6 64 bit").
+4. OpenMS version (e.g. "OpenMS 1.11.1", "Revision 63082 from the SVN repository").
+5. OpenMS architecture ("32 bit" or "64 bit")
+
+Please provide files that we need to reproduce the bug (e.g. `TOPP INI` files, data files — usually mzML) via a download
+link, via the mailing list or by directly contacting one of the developers.
diff --git a/docs/additional-resources/write-and-label-github-issues.md b/docs/additional-resources/write-and-label-github-issues.md
new file mode 100644
index 00000000..f1f43192
--- /dev/null
+++ b/docs/additional-resources/write-and-label-github-issues.md
@@ -0,0 +1,24 @@
+Write and Label GitHub Issues
+=============================
+
+## Create an Issue
+
+To create an issue:
+
+1. Go to the [OpenMS codebase](https://github.com/OpenMS/OpenMS).
+2. Submit an [issue](https://github.com/OpenMS/OpenMS/issues/new).
+
+The issue will be listed under **Issues**.
+
+## Label an Issue
+
+To label an issue:
+
+1. On the right of the screen, select the cog icon under **Labels**.
+2. Choose a label from the list. Normally, an issue can have one or more of the following labels:
+ - **defect**: A defect refers to a bug in OpenMS. This is a high priority issue.
+ - **enhancement**: An enhancement refers to a feature idea to enhance the current OpenMS code. This is a medium
+ priority issue.
+ - **task**: A task refers to a single piece of work that a developer can undertake. This is a medium priority issue.
+ - **refactoring**: A refactoring issue refers to a suggestion to streamline the code without changing how the code function.
+ - **question**: A question could trigger to a discussion about tools, parameters and scientific tasks.
diff --git a/docs/advanced-resources/custom-compilation.md b/docs/advanced-resources/custom-compilation.md
new file mode 100644
index 00000000..0cba8b7e
--- /dev/null
+++ b/docs/advanced-resources/custom-compilation.md
@@ -0,0 +1,35 @@
+Custom Compilation of OpenMS
+===========================
+
+To compile with self built compilers and non default standard libraries, follow listed steps.
+
+To choose any specific compiler, instead of the system default, add the whole path to these options for the cmake call:
+
+```{tab} GCC
+
+`cmake -DCMAKE_C_COMPILER=/path/to/c-compiler/binary/gcc -DCMAKE_CXX_COMPILER=/path/to/c++-compiler/binary/g++`
+ ```
+```{tab} Clang
+
+`cmake -DCMAKE_C_COMPILER=/path/to/c-compiler/binary/clang -DCMAKE_CXX_COMPILER=/path/to/c++-compiler/binary/clang++`
+```
+
+To compile OpenMS with clang and a specific GCC stdlib, instead of the system default one:
+
+Use this cmake option to specify an additional compiling option for clang:
+
+```bash
+cmake -DMY_CXX_FLAGS="--gcc-toolchain=/path/to/gcc"
+```
+
+with the path to the top gcc directory (containing the directory lib64) to the cmake call.
+
+```{warning}
+This combination does not work for all versions of clang and gcc.
+- Clang 9.0.0 and GCC 4.8.5 stdlib does not work!
+- Clang 9.0.0 and GCC 9.2.0 stdlib does not work!
+- Clang 9.0.0 and GCC 8.3.0 stdlib compiles, but some tests fail.
+- Clang 6.0.0 and GCC 7.4.0 stdlib (Ubuntu 18.04 default versions) works
+```
+
+
diff --git a/docs/contact-us.md b/docs/contact-us.md
new file mode 100644
index 00000000..6d3cc82c
--- /dev/null
+++ b/docs/contact-us.md
@@ -0,0 +1,15 @@
+Contact Us
+=========
+
+Join us on [Discord](https://discord.gg/WxynEEsf5X)!
+
+You can also contact us:
+
+1. On the user and contributor real time [Gitter chat](https://gitter.im/OpenMS/OpenMS).
+2. Drop us an email at user support [open-ms-general](https://sourceforge.net/projects/open-ms/lists/open-ms-general)
+ mailing list.
+3. To stay updated of new versions of OpenMS and releases, subscribe to [openms-announcements mailing list](https://sourceforge.net/projects/open-ms/lists/open-ms-announcements).
+4. To report a new bug, create an issue on GitHub [OpenMS](https://github.com/OpenMS/OpenMS/issues) repository.
+
+
+Say hi on [OpenMS Twitter](https://twitter.com/openmsteam)!
diff --git a/docs/downloads.md b/docs/downloads.md
new file mode 100644
index 00000000..647f8de7
--- /dev/null
+++ b/docs/downloads.md
@@ -0,0 +1,42 @@
+# OpenMS Installers
+
+| Platform | Name | SHA256 Hash |
+|----------|------|-------------|
+| Windows | [OpenMS-2.8.0-Win64.exe](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/2.8.0/OpenMS-2.8.0-Win64.exe) | `d203985c7042b885ac1085c30a2d9f36d7609b47`|
+| macOS | [OpenMS-2.8.0-macOS.dmg](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/2.8.0/OpenMS-2.8.0-macOS.dmg) | `d203985c7042b885ac1085c30a2d9f36d7609b47` |
+| GNU/Linux | [OpenMS-2.8.0-Debian-Linux-x86_64.deb](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/2.8.0/OpenMS-2.8.0-Debian-Linux-x86_64.deb) | `d203985c7042b885ac1085c30a2d9f36d7609b47` |
+| Source | [OpenMS-2.8.0-src.tar.gz](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/2.8.0/OpenMS-2.8.0-src.tar.gz) | `d203985c7042b885ac1085c30a2d9f36d7609b47` |
+
+
+# Workflows
+
+| Workflow | Description | Download Link |
+|----------|-------------|---------------|
+|`ProteomicsLFQ_tool_and_MSstats_postprocessing` | Label-free identification and quantification using the comet search engine, the ProteomicsLFQ tool and statistical down-stream processing using MSstats. Compared to the other proteomics LFQ workflows, it is less complex as it combines quantification and inference steps in a single ProtemicLFQ tool. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/ProteomicsLFQ_tool_and_MSstats_postprocessing.knwf?raw=true) |
+|`basic_peptide_identification` | Label-free identification using the omssa search engine. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/basic_peptide_identification.knwf?raw=true) |
+|`DIAMetAlyzer` | Metabolomics assay library construction with decoy generation from DDA data and targeted DIA analysis using OpenSWATH and pyprophet for statistical validation. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/DIAMetAlyzer.knwf?raw=true) |
+|`Identification_quantification_with_inference_isobaric_epifany_MSstatsTMT` | Identification and quantification for isobaric experiments using MSGFPlus as search engine, epifany for inference and MSstatsTMT for statistical down-stream analysis. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Identification_quantification_with_inference_isobaric_epifany_MSstatsTMT.knwf?raw=true) |
+|`labelfree_with_protein_quantification` | Label-free with protein quantification steps implemented using individual OpenMS tools | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/labelfree_with_protein_quantification.knwf?raw=true) |
+|`Metabolite_Adduct_Grouping` | Quantification and identification via accurate mass based on multiple adduct grouping steps (adducts, neutral losses). | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Metabolite_Adduct_Grouping.knwf?raw=true) |
+|`Metabolite_DeNovoID` | Quantification and identification via adduct grouping and de-novo identification using SIRIUS/CSI:FingerID. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Metabolite_DeNovoID.knwf?raw=true) |
+|`Metabolite_ID` | Quantification and identification via accurate mass based with downstream processing and visualisation. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Metabolite_ID.knwf?raw=true) |
+|`Metabolite_SpectralID` | Identification via spectral library search for small molecules. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Metabolite_SpectralID.knwf?raw=true) |
+|`MSstats_statPostProcessing_iPRG2015` | Post processing workflow for using MSstats based on "Example_OneTool_ProteomicsLFQ_MSstats.knwf" | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/MSstats_statPostProcessing_iPRG2015.knwf?raw=true) |
+|`MSstatsTMT` | Post processing workflow for using MSstatsTMT based on "Identification_quantification_with_inference_isobaric_epifany_MSstatsTMT". | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/MSstatsTMT.knwf?raw=true) |
+|`OpenSWATH` | Targeted extraction and scoring of transitions in DIA data based on an (iRT) assay library. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/OpenSWATH.knwf?raw=true) |
+|`Phosphoproteomics_ID` | Identification of Phosphorilation sites. | [Download](https://github.com/OpenMS/Tutorials/blob/master/Workflows/Phosphoproteomics_ID.knwf?raw=true) |
+
+# OpenMS Releases
+
+| Release | Installers |
+|--------------------------------------------------------|-------------|
+| Stable release | [Archive Link](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/) |
+| Release candidates | [Archive Link](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/RC/) |
+| Nightly release | [Archive Link](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/nightly/) |
+
+# Other Resources
+
+| Name | Description | Download Link |
+|------|-------------|---------------|
+| Schemas | Documented schemas of the OpenMS formats | [Download](https://github.com/OpenMS/OpenMS/tree/develop/share/OpenMS/SCHEMAS)|
+| iPRG2016 data | Dataset mxMLs, Fasta database, Identification file (idXML), Big Data (idXML) | [Download](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Tutorials/Data/iPRG2016/) |
diff --git a/docs/faqs/contributor-faq.md b/docs/faqs/contributor-faq.md
new file mode 100644
index 00000000..aaf96ebe
--- /dev/null
+++ b/docs/faqs/contributor-faq.md
@@ -0,0 +1,200 @@
+Contributor FAQ
+===============
+
+The following contains answers to typical questions from contributors about OpenMS.
+
+## General
+
+The following section provides general information to new contributors.
+
+### I am new to OpenMS. What should I do first?
+
+* Check out the development version of OpenMS (see OpenMS [releases and installers](downloads/releases.md)).
+* Build OpenMS according to the installation instructions.
+* Read the [OpenMS Coding Conventions](https://github.com/OpenMS/OpenMS/wiki/Coding-conventions).
+* Read the [OpenMS Tutorial](../tutorials/user-tutorial.md).
+* Create a GitHub account.
+* Subscribe to the [open-ms-general](https://sourceforge.net/projects/open-ms/lists/open-ms-general) or [contact us](../contact-us.md).
+
+### What is the difference between an OpenMS tool and util?
+
+A tool starts its lifecycle in `UTILS` and may exist without being thoroughly tested. Tools may be promoted from `UTILS`
+to `TOOLS` if they are stable enough, are fully tested, fully documented, and a test workflow exists.
+
+### I have written a class for OpenMS. What should I do?
+
+Follow the [OpenMS coding conventions]().
+
+Coding style (brackets, variable names, etc.) must conform to the conventions.
+
+* The class and all the members must be documented thoroughly.
+* Check your code with the tool `tools/checker.php`. Call `php tools/checker.php` for detailed instructions.
+
+Please open a pull request and follow the [pull request guidelines](pull-request-checklist.md).
+
+## Troubleshooting
+
+The following section provides information about how to troubleshoot common OpenMS issues.
+
+### OpenMS complains about boost not being found
+
+`CMake` got confused. Set up a new build directory and try again. Build from source, deleting the `CMakeCache.txt`
+and `cmake` directory might help.
+
+## Build System
+
+The following questions are related to the build system.
+
+### What is CMake?
+
+`CMake` builds BuildSystems for different platforms, e.g. VisualStudio Solutions on Windows, Makefiles on Linux etc.
+This allows us to define in one central location (namely `CMakeLists.txt`) how OpenMS is build and have the platform
+specific stuff handled by `CMake`. View the [cmake website](http://www.cmake.org) for more information.
+
+### How do I use CMake?
+
+See Installation instructions for your platform.
+In general, call `CMake(.exe)` with some parameters to create the native build-system.
+Afterwards, (but usually) don't have to edit the current configuration using a GUI named `ccmake`
+(or `CMake-GUI` in Windows), which ships with `CMake`).
+
+```{note}
+Whenever `ccmake` is mentioned in this document, substitute this by `CMake-GUI` if your OS is Windows. Edit
+the `CMakeCache.txt` file directly.
+```
+
+### How do I generate a build-system for Eclipse, KDevelop, CodeBlocks etc?
+
+Type `cmake` into a console. This will list the available code generators available on your platform, pass them to `CMake`
+using the `-G` option.
+
+### How do I add a new class to the build system?
+
+1. Create the new class in the corresponding sub-folder of the sub-project. The header has to be created in
+ `src//include/OpenMS` and the cpp file in `src//source`, e.g.,
+ `src/openms/include/OpenMS/FORMAT/NewFileFormat.h` and `src/openms/source/FORMAT/NewFileFormat.cpp`.
+2. Add both to the respective `sources.cmake` file in the same directory (e.g., `src/openms/source/FORMAT/` and
+ `src/openms/include/OpenMS/FORMAT/`).
+3. Add the corresponding class test to `src/tests/class_tests//`
+ (e.g., `src/tests/class_tests/openms/source/NewFileFormat_test.cpp`).
+4. Add the test to the `executables.cmake` file in the test folder (e.g., `src/tests/class_tests/openms/executables.cmake`).
+5. Add them to git by using the command `git add`.
+
+### How do I add a new directory to the build system?
+
+1. Create two new `sources.cmake` files (one for `src//include/OpenMS/MYDIR`, one for
+ `src//source/MYDIR`), using existing `sources.cmake` files as template.
+2. Add the new `sources.cmake` files to `src//includes.cmake`
+3. If you created a new directory directly under `src/openms/source`, then have a look at `src/tests/class_tests/openms/executables.cmake`.
+4. Add a new section that makes the unit testing system aware of the new (upcoming) tests.
+5. Look at the very bottom and augment `TEST_executables`.
+6. Add a new group target to `src/tests/class_tests/openms/CMakeLists.txt`.
+
+## Debugging
+
+The following section provides information about how to debug your code.
+
+### How do I run a single test?
+
+Execute an OpenMS class test using the CTest regular expressions:
+
+```bash
+
+$ ctest -V -R "^_test"
+
+# To build a class test, call the respective make target in ./source/TEST:
+
+$ make _test
+```
+To run a TOPP test, use:
+
+```bash
+
+$ ctest -V -R "TOPP_"
+```
+
+To build the tool, use:
+
+```bash
+$ make
+```
+### How do I debug uncaught exceptions?
+
+Dump a core if an uncaught exception occurs, by setting the environment variable `OPENMS_DUMP_CORE`.
+
+Each time an uncaught exception occurs, the `OPENMS_DUMP_CORE` variable is checked and a segmentation fault is caused,
+if it is set.
+
+### (Linux) How can I set breakpoints in gdb to debug OpenMS?
+
+Debug the TOPPView application to stop at line 341 of SpectrumMDIWindow.C.
+
+1. Enter the following in your terminal:
+
+ ```bash
+ Run gdb:
+ shell> gdb TOPPView
+```
+
+2. Start the application (and close it):
+
+ ```bash
+ gdb> run [arguments]
+```
+
+3. Set the breakpoint:
+ ```bash
+ gdb> break SpectrumMDIWindow.C:341
+```
+
+4. Start the application again (with the same arguments):
+
+ ```bash
+ gdb> run
+ ```
+
+## Doxygen Documentation
+
+### Where can I find the definition of the main page?
+
+Find a definition of the main page [here](https://github.com/OpenMS/OpenMS/edit/develop/doc/doxygen/public/Main.doxygen).
+
+### Where can I add a new module?
+
+Add a new module [here](https://github.com/OpenMS/OpenMS/edit/develop/doc/doxygen/public/Modules.doxygen).
+
+
+### How is the command line documentation for TOPP/UTILS tools created?
+
+The program `OpenMS/doc/doxygen/parameters/TOPPDocumenter.cpp` creates the command line documentation for all classes
+that are included in the static `ToolHandler.cpp` tools list. It can be included in the documentation using the following `doxygen` command:
+
+`@verbinclude TOPP_.cli`
+
+Test if everything worked by calling `make doc_param_internal`. The command line documentation is written to
+`OpenMS/doc/doxygen/parameters/output/`.
+
+### What are the important files for adding a new tutorial section?
+
+View the following OpenMS tutorials:
+
+* `OpenMS/doc/OpenMS_tutorial/refman_overwrite.tex.in` (for PDF tutorials)
+* `OpenMS/doc/doxygen/public/OpenMS_Tutorial_html.doxygen` (for html tutorials)
+
+View the following TOPP and TOPPView tutorials:
+
+* `OpenMS/doc/TOPP_tutorial/refman_overwrite.tex.in` (for PDF tutorials)
+* `OpenMS/doc/doxygen/public/TOPP_Tutorial_html.doxygen` (for html tutorials)
+
+## Bug Fixes
+
+### How do I contribute to a bug fix?
+
+To contribute to a bug fix:
+
+1. Submit the bug as a GitHub issue.
+2. Create a feature branch (e.g. `feature/fix_missing_filename_issue_615`) from your (up-to-date) develop branch in your fork of OpenMS.
+3. Fix the bug and add a test.
+4. Create a pull request for your branch.
+5. After approval and merge make sure the issue is closed.
+
diff --git a/docs/faqs/developer-faq.md b/docs/faqs/developer-faq.md
new file mode 100644
index 00000000..81b46548
--- /dev/null
+++ b/docs/faqs/developer-faq.md
@@ -0,0 +1,399 @@
+Developer FAQ
+=============
+
+The following contains answers to typical questions from developers about OpenMS.
+
+## General
+
+The following section provides general information to new contributors.
+
+### I am new to OpenMS. What should I do first?
+
+* Check out the development version of OpenMS (see website).
+* Build OpenMS by following the [installation instructions]() or [from source](../installations/build-openms-from-source.md).
+* Read the [OpenMS Coding Conventions]()
+* Read the [OpenMS Tutorial](../tutorials/user-tutorial.md).
+* Create a GitHub account.
+* Subscribe to the [open-ms-general](https://sourceforge.net/projects/open-ms/lists/open-ms-general) or [contact-us](../contact-us.md).
+
+### What is the difference between an OpenMS tool and util?
+
+A tool starts its lifecycle in `UTILS` and may exist without being thoroughly tested. Tools may be promoted from `UTILS`
+to `TOOLS` if they are stable enough, are fully tested, fully documented, and a test workflow exists.
+
+### I have written a class for OpenMS. What should I do?
+
+Follow the [OpenMS coding conventions]().
+
+Coding style (brackets, variable names, etc.) must conform to the conventions.
+
+* The class and all the members should be properly documented.
+* Check your code with the tool `tools/checker.php`. Call `php tools/checker.php` for detailed instructions.
+
+Please open a pull request and follow the [pull request guidelines](pull-request-checklist.md).
+
+### Can I use QT designer to create GUI widgets?
+
+Yes. Create a class called `Widget: Create .ui-File` with `QT designer` and store it as `Widget.ui.`, add the class to
+`sources.cmake`. From the .ui-File the file `include/OpenMS/VISUAL/UIC/ClassTemplate.h` is generated by the build system.
+
+```{note}
+Do not check in this file, as it is generated automatically when needed.
+```
+
+Derive the class `Widget` from `WidgetTemplate`. For further details, see the `Widget.h` and `Widget.cpp` files.
+
+### Can the START_SECTION-macro not handle template methods that have two or more arguments?
+
+Insert round brackets around the method declaration.
+
+### Where can I find the binary installers created?
+
+View the binary installers at the [build archive](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/nightly/).
+Please verify the creation date of the individual installers, as there may have been an error while creating the installer.
+
+## Troubleshooting
+
+The following section provides information about how to troubleshoot common OpenMS issues.
+
+### OpenMS complains about boost not being found but I'm sure its there
+
+`CMake` got confused. Set up a new build directory and try again. If you build from source (not recommended), deleting
+the `CMakeCache.txt` and `cmake` directory might help.
+
+## Build System
+
+The following questions are related to the build system.
+
+### What is CMake?
+
+`CMake` builds BuildSystems for different platforms, e.g. VisualStudio Solutions on Windows, Makefiles on Linux etc.
+This allows to define in one central location (namely `CMakeLists.txt`) how OpenMS is build and have the platform specific
+stuff handled by `CMake`.
+
+View the [cmake website](http://www.cmake.org) for more information.
+
+### How do I use CMake?
+
+See Installation instructions for your platform.
+In general, call `CMake(.exe)` with some parameters to create the native build-system.
+
+```{tip}
+whenever `ccmake` is mentioned in this document, substitute this by `CMake-GUI` if your OS is Windows. Edit the
+`CMakeCache.txt` file directly.
+```
+
+### How do I generate a build-system for Eclipse, KDevelop, CodeBlocks etc?
+
+Type `cmake` into a console. This will list the available code generators available on your platform; use them with
+`CMake` using the `-G` option.
+
+### What are user definable CMake cache variables?
+
+They allow the user to pass options to `CMake` which will influence the build system. The most important option which
+should be given when calling `CMake.exe` is:
+
+`CMAKE_FIND_ROOT_PATH`, which is where `CMake` will search for additional libraries if they are not found in the default
+system paths. By default we add `OpenMS/contrib`.
+
+If you have installed all libraries on your system already, there is no need to change `CMAKE_FIND_ROOT_PATH`. For
+`contrib` libraries, set the variable `CMAKE_FIND_ROOT_PATH`.
+
+On Windows, `contrib` folder is required, as there are no system developer packages. To pass this variable to
+`CMake` use the `-D` switch e.g. `cmake -D CMAKE_FIND_ROOT_PATH:PATH="D:\\somepath\\contrib"`.
+
+Everything else can be edited using `ccmake` afterwards.
+
+The following options are of interest:
+
+- `CMAKE_BUILD_TYPE` To build Debug or Release version of OpenMS. Release is the default.
+- `CMAKE_FIND_ROOT_PATH` The path to the `contrib` libraries.
+ ```{tip}
+ Provide more then one value here (e.g., `-D CMAKE_FIND_ROOT_PATH="/path/to/contrib;/usr/"` will search in your
+ `contrib` path and in `/usr` for the required libraries)
+ ```
+- `STL_DEBUG` Enables STL debug mode.
+- `DB_TEST` (deprecated) Enables database testing.
+- `QT_DB_PLUGIN` (deprecated) Defines the db plugin used by Qt.
+
+View the description for each option by calling `ccmake`.
+
+### Can I use another solver other than GLPK?
+
+Other solvers can be used, but by default, the build system only links to GLPK (this is how OpenMS binary packages must
+be built). To to use another solver, use `cmake ... -D USE_COINOR=1 ....` and refer to the documentation of the
+`LPWrapper` class.
+
+### How do I switch to debug or release configuration?
+
+For Makefile generators (typically on Linux), set the `CMAKE_BUILD_TYPE` variable to either Debug or Release by
+calling `ccmake`. For Visual Studio, this is not necessary as all configurations are generated and choose the one you
+like within the IDE itself. The 'Debug' configuration enabled debug information. The 'Release' configuration disables
+debug information and enables optimisation.
+
+### I changed the `contrib` path, but re-running `CMake` won't change the library paths?
+
+Once a library is found and its location is stored in a cache variable, it will only be searched again if the
+corresponding entry in the cache file is set to false.
+
+```{warning}
+If you delete the `CMakeCache.txt`, all other custom settings will be lost.
+```
+
+The most useful targets will be shown to you by calling the targets target, i.e. make targets.
+
+### `CMake` can't seem to find a `Qt` library (usually `QtCore`). What now?
+
+`CMake` finds `QT` by looking for `qmake` in your `PATH` or for the Environment Variable `QTDIR`. Set these accordingly.
+
+Make sure there is no second installation of Qt (especially the MinGW version) in your local environment.
+```{warning}
+This might lead ``CMake`` to the wrong path (it's searching for the ``Qt*.lib`` files).
+You should only move or delete the offending `Qt` version if you know what you are doing!
+```
+
+A save workaround is to edit the `CMakeCache` file (e.g. via `ccmake`) and set all paths relating to `QT`
+(e.g. `QT_LIBRARY_DIR`) manually.
+
+### (Windows) What version of Visual Studio should I use?
+
+It is recommended to use the latest version. Get the latest `CMake`, as its generator needs to support your VS. If
+your VS is too new and there is no `CMake` for that yet, you're gonna be faced with a lot of conversion issues.
+This happens whenever the Build-System calls `CMake` (which can be quite often, e.g., after changes to `CMakeLists.txt`).
+
+### How do I add a new class to the build system?
+
+1. Create the new class in the corresponding sub-folder of the sub-project. The header has to be created in `src//include/OpenMS` and the `.cpp` file in `src//source`, e.g., `src/openms/include/OpenMS/FORMAT/NewFileFormat.h` and `src/openms/source/FORMAT/NewFileFormat.cpp`.
+2. Add both to the respective `sources.cmake` file in the same directory (e.g., `src/openms/source/FORMAT/` and `src/openms/include/OpenMS/FORMAT/`).
+3. Add the corresponding class test to `src/tests/class_tests//` (e.g., `src/tests/class_tests/openms/source/NewFileFormat_test.cpp`).
+4. Add the test to the `executables.cmake` file in the test folder (e.g., `src/tests/class_tests/openms/executables.cmake`).
+5. Add them to git by using the command `git add`.
+
+### How do I add a new directory to the build system?
+
+1. Create two new `sources.cmake` files (one for `src//include/OpenMS/MYDIR`, one for `src//source/MYDIR`), using existing `sources.cmake` files as template.
+2. Add the new `sources.cmake` files to `src//includes.cmake`
+3. If you created a new directory directly under `src/openms/source`, then have a look at `src/tests/class_tests/openms/executables.cmake`.
+4. Add a new section that makes the unit testing system aware of the new (upcoming) tests.
+5. Look at the very bottom and augment `TEST_executables`.
+6. Add a new group target to `src/tests/class_tests/openms/CMakeLists.txt`.
+
+### How can I speed up the compile process of OpenMS?
+
+To speed up the compile process of OpenMS, use several threads. If you have several processors/cores, build OpenMS
+classes/tests and `TOPP` tools in several threads. On Linux, use the `make option -j: make -j8 OpenMS TOPP test_build`.
+
+On Windows, Visual Studio solution files are automatically build with the `/MP` flag, such that Visual Studio uses all
+available cores of the machine.
+
+## Release
+
+View [preparation of a new OpenMS release](https://github.com/OpenMS/OpenMS/wiki/Preparation-of-a-new-OpenMS-release#release_developer) to learn more about contributing to releases.
+
+
+## Working in Integrated Development Environments (IDEs)
+
+### Why are there no `source/TEST` and `source/APPLICATIONS/TOPP|UTILS` folder?
+
+All source files added to an IDE are associated with their targets. Find the source files for each test within
+its own subproject. The same is true for the `TOPP` and `UTILS` classes.
+
+### I'm getting the error "Error C2471: cannot update program database"
+
+This is a bug in Visual Studio and there is a [bug fix](http://code.msdn.microsoft.com/KB946040) Only apply it if you
+encounter the error. The bug fix might have unwanted side effects!
+
+### Visual Studio can't read the clang-format file.
+
+Depending on the Visual Studio version it might get an error like `Error while formating with ClangFormat`.
+This is because Visual Studio is using an outdated version of clang-format. Unfortunately there is no easy way to update
+this using Visual Studio itself. There is a plugin provided by LLVM designed to fix this problem, but the plugin doesn't
+work with every Visual Studio version. In that case, update clang-format manually using the pre-build clang-format binary.
+Both the binary and a link to the plugin can be found [here](https://llvm.org/builds/).
+To update clang-format download the binary and exchange it with the clang-format binary in your Visual Studio folder.
+For Visual Studio 17 and 19 it should be located at: `C:\Program Files (x86)\Microsoft Visual Studio\2019\Community\VC\Tools\Llvm\bin`.
+
+### The indexer gets stuck at some file which ``#includes seqan``
+
+It seems that SeqAn code is just too confusing for older eclipse C++ indexers. You should upgrade to eclipse galileo
+(CDT 6.0.x). Also, increase the available memory limit in `eclipse.ini`, e.g. `-Xmx1024m` for one gig.
+
+### The parser is confused after OPENMS_DLLAPI and does not recognize standard C++ headers
+
+Go to ``Project -> Properties -> C/C++ Include Paths and Preprocessor Symbols -> Add Preprocessor symbol -> "OPENMS_DLLAPI="``.
+This tells eclipse that the macro is defined empty. In the same dialog add an external include path to
+e.g. ``/usr/include/c++/4.3.3/``, etc. The issue with C++ headers was fixed in the latest galileo release.
+
+Hints to resolve the OPENMS_DLLAPI issue using the ``cmake`` generator are welcome!
+
+## Debugging
+
+The following section provides information about how to debug your code.
+
+### How do I debug uncaught exceptions?
+
+Dump a core if an uncaught exception occurs, by setting the environment variable `OPENMS_DUMP_CORE`.
+
+Each time an uncaught exception occurs, the `OPENMS_DUMP_CORE` variable is checked and a segmentation fault is caused,
+if it is set.
+
+### (Linux) Why is no core dumped, although a fatal error occured?
+
+The `ulimit -c` unlimited command. It sets the maximum size of a core to unlimited.
+
+```{warning}
+We observed that, on some systems, no core is dumped even if the size of the core file is set to unlimited. We are not
+sure what causes this problem.
+```
+
+### (Linux) How can I set breakpoints in gdb to debug OpenMS?
+
+Imagine you want to debug the TOPPView application and you want it to stop at line 341 of SpectrumMDIWindow.C.
+
+1. Enter the following in your terminal:
+
+ ```bash
+ Run gdb:
+ shell> gdb TOPPView
+```
+
+2. Start the application (and close it):
+
+ ```bash
+ gdb> run [arguments]
+```
+3. Set the breakpoint:
+ ```bash
+ gdb> break SpectrumMDIWindow.C:341
+```
+4. Start the application again (with the same arguments):
+
+ ```bash
+ gdb> run
+ ```
+
+### How can I find out which shared libraries are used by an application?
+
+Linux: Use `ldd`.
+
+Windows (Visual studio console): See [Dependency Walker](http://www.dependencywalker.com/) (use x86 for 32 bit builds
+and the x64 version for 64bit builds. Using the wrong version of depends.exe will give the wrong results) or
+``dumpbin /DEPENDENTS OpenMS.dll``.
+
+### How can I get a list of the symbols defined in a (shared) library or object file?
+
+Linux: Use `nm `.
+
+Use `nm -C` to switch on demangling of low-level symbols into their C++-equivalent names. `nm` also accepts .a and .o files.
+
+Windows (Visual studio console): Use ``dumpbin /ALL ``.
+
+Use dumpbin on object files (.o) or (shared) library files (.lib) or the DLL itself e.g. `dumpbin /EXPORTS OpenMS.dll`.
+
+## Cross-platform thoughts
+
+OpenMS runs on three major platforms.. Here are the most prominent causes of "it runs on Platform A, but not on B. What now?"
+
+### Reading or writing binary files
+
+Reading or writing binary files causes different behaviour. Usually Linux does not make a difference between text-mode
+and binary-mode when reading files. This is quite different on Windows as some bytes are interpreted as `EOF`, which lead
+might to a premature end of the reading process.
+
+If reading binary files, make sure that you explicitly state that the file is binary when opening it.
+
+During writing in text-mode on Windows a line-break (`\n`) is expanded to (`\r\n`). Keep this in mind or use the
+`eol-style` property of subversion to ensure that line endings are correctly checked out on non-Windows systems.
+
+### Paths and system functions
+
+Avoid hardcoding e.g.`String tmp_dir = "/tmp";`. This will fail on Windows. Use Qt's `QDir` to get a path to the systems
+temporary directory if required.
+
+Avoid names like uname which are only available on Linux.
+
+When working with files or directories, it is usually safe to use "/" on all platforms. Take care of spaces in directory
+names though. Quote paths if they are used in a system call to ensure that the subsequent interpreter
+takes the spaced path as a single entity.
+
+
+## Doxygen Documentation
+
+### Where can I find the definition of the main page?
+
+Find a definition of the main page [here](https://github.com/OpenMS/OpenMS/edit/develop/doc/doxygen/public/Main.doxygen).
+
+### Where can I add a new module?
+
+Add a new module [here](https://github.com/OpenMS/OpenMS/edit/develop/doc/doxygen/public/Modules.doxygen).
+
+### How is the parameter documentation for classes derived from DefaultParamHandler created?
+
+Add your class to the program ``OpenMS/doc/doxygen/parameters/DefaultParamHandlerDocumenter.cpp``. This program generates
+a html table with the parameters. This table can then be included in the class documentation using the following
+`doxygen` command:`@htmlinclude OpenMS_.parameters`.
+
+```{note}
+Parameter documentation is automatically generated for `TOPP/UTILS` included in the static `ToolHandler.cpp` tools list.
+```
+
+To include TOPP/UTILS parameter documentation use following `doxygen` command:
+
+
+`@htmlinclude TOPP_.parameters`
+
+or
+
+`@htmlinclude UTILS_.parameters`
+
+Test if everything worked by calling `make doc_param_internal`. The parameters documentation is written to
+`OpenMS/doc/doxygen/parameters/output/`.
+
+### How is the command line documentation for TOPP/UTILS tools created?
+
+The program `OpenMS/doc/doxygen/parameters/TOPPDocumenter.cpp` creates the command line documentation for all classes
+that are included in the static `ToolHandler.cpp` tools list. It can be included in the documentation using the following `doxygen` command:
+
+`@verbinclude TOPP_.cli`
+
+Test if everything worked by calling `make doc_param_internal`. The command line documentation is written to
+`OpenMS/doc/doxygen/parameters/output/`.
+
+## Bug Fixes
+
+### How to contribute a bug fix?
+
+Read [contributor quickstart guide](../guides/contributors-quickstart-guide.md).
+
+### How can I profile my code?
+
+IBM's profiler, available for all platforms (and free for academic use): Purify(Plus) and/or Quantify.
+
+Windows: this is directly supported by Visual Studio (Depending on the edition: Team and above). Follow their documentation.
+
+Linux:
+
+1. Build OpenMS in debug mode (set `CMAKE_BUILD_TYPE` to `Debug`).
+2. Call the executable with valgrind: `valgrind –tool=callgrind`.
+ ```{warning}
+ Other processes running on the same machine can influence the profiling. Make sure your application gets enough
+ resources (memory, CPU time).
+ ```
+3. Start and stop the profiling while the executable is running e.g. to skip initialization steps:
+4. Start valgrind with the option `–instr-atstart=no`.
+5. Call `callgrind -i [on|off]` to start/stop the profiling.
+6. The output can be viewed with `kcachegrind callgrind.out`.
+
+### (Linux) How do I check my code for memory leaks?
+
+* Build OpenMS in debug mode (set ``CMAKE_BUILD_TYPE`` to ``Debug``).
+* Call the executable with ``valgrind: valgrind --suppressions=OpenMS/tools/valgrind/openms_external.supp –leak-check=full ``.
+
+Common errors are:
+
+* ``'Invalid write/read ...'`` - Violation of container boundaries.
+* ``'... depends on uninitialized variable'`` - Uninitialized variables:
+* ``'... definitely lost'`` - Memory leak that has to be fixed
+* ``'... possibly lost'`` - Possible memory leak, so have a look at the code
+
+For more information see the [`valgrind` documentation](http://valgrind.org/docs/manual/) .
diff --git a/docs/glossary.md b/docs/glossary.md
new file mode 100644
index 00000000..764ae707
--- /dev/null
+++ b/docs/glossary.md
@@ -0,0 +1,187 @@
+OpenMS Glossary
+==============
+
+A glossary of common terms used throughout OpenMS documentation.
+
+```{glossary}
+
+OpenMS API
+ [OpenMS API Reference](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html).
+ The object-oriented OpenMS core library contains over 1,300 classes and is built on modern C++ infrastructure
+ with native compiler support on Windows, Linux and macOS. The classes are representing core concepts in mass
+ spectrometry as well as the corresponding ontologies defined by the Human Proteome Organization Proteomics
+ Standard Initiative (HUPO-PSI).
+
+Workflow
+ A set of over 185 different tools for common mass spectrometric tasks can be accessed by routine users through
+ the KNIME, and Galaxy workflow systems.
+
+SILAC
+ **S**table **I**sotope **L**abeling by/with **A**mino acids in **C**ell culture is a technique based on mass
+ spectrometry that detects differences in protein abundance among samples using non-radioactive isotopic labeling.
+
+iTRAQ
+ **I**sobaric **T**ags for **R**elative and **A**bsolute **A**uantitation (iTRAQ) is an isobaric labeling method
+ used in quantitative proteomics by tandem mass spectrometry to determine the amount of proteins from different
+ sources in a single experiment.
+
+TMT
+ A **T**andem **M**ass **T**ag (TMT) is a chemical label that facilitates sample multiplexing in mass spectrometry
+ ({term}`MS`)-based quantification and identification of biological macromolecules such as proteins, {term}`peptides`
+ and nucleic acids.
+
+SRM
+ **S**elected **R**eaction **M**onitoring (SRM), also called Multiple reaction monitoring, (MRM), is a method used
+ in tandem mass spectrometry in which an ion of a particular mass is selected in the first stage of a tandem mass
+ spectrometer and an ion product of a fragmentation reaction of the precursor ions is selected in the second mass
+ spectrometer stage for detection.
+
+SWATH
+ SWATH-mass spectrometry consists of data-independent acquisition and a targeted data analysis strategy that aims
+ to maintain the favorable quantitative characteristics (accuracy, sensitivity, and selectivity) of targeted
+ proteomics at large scale.
+
+KNIME
+ **K**onstanz **I**nformation **M**iner, is a free and open-source data analytics, reporting and integration platform.
+
+LC-MS
+ [Liquid Chromatography(LC)](introduction.md#liquid-chromatography-lc) and [Mass Spectrometry(MS)](introduction.md#mass-spectrometry).
+
+Peptides
+ A short chain of amino acids.
+
+Octadecyl(C18)
+ An alkyl radical C(18)H(37) derived from an octadecane by removal of one hydrogen atom.
+
+Mass
+ Mass is a measure of the amount of matter that an object contains. In comparison to often used term weight, which is
+ a measure of the force of gravity on that object.
+
+Ion
+ Any {term}`atom` or group of atoms that bears one or more positive or negative electrical charges. Positively charged are
+ cations, negavtively charged anions.
+
+Atom
+ An atom is the smallest unit of ordinary matter that forms a chemical element.
+
+Electrospray ionization
+ A technique used in mass spectrometry to produce ions using an electrospray in which a high voltage is applied to a
+ liquid to create an {term}`aerosol`.
+
+Aerosol
+ An aerosol is a suspension of fine solid particles or liquid droplets in air or another gas.
+
+Time-of-flight (TOF)
+ A measurement of the time taken by an object, particle of wave (be it acoustic, electromagnetic, e.t.c) to travel a
+ distance through a medium.
+
+Quadrupole mass filters
+ A mass filter allowing one mass channel at a time to reach the detector as the mass range is scanned.
+
+Orbitrap analyzers
+ In mass spectrometry, an ion trap mass analyzer consisting of an outer barrel-like electrode and a coaxial inner
+ spindle-like electrode that traps ions in an orbital motion around the spindle.
+ A high resoltion mass spectrometry analyzer.
+
+MS(1)
+ First stage to get a spectra. A sample is injected into the mass spectrometer, ionized, accelerated and analyzed by
+ mass spectrometry.
+
+MS(2)
+ Ions from MS1 spectra are then selectively fragmented and analyzed by a second stage of mass spectrometry (MS2) to
+ generate the spectra for the ion fragments.
+
+MS/MS
+ Tandem mass spectrometry, MS^2^, a technique where two or more mass analyzers are coupled together using an additional
+ reaction step to increase their abilities to analyse chemical samples.
+
+Collision-induced dissociation (CID)
+ A mass spectrometry technique to induce fragmentation of selected ions in the gas phase. Also known as Collision
+ induced dissociation.
+
+TOPP
+ The OpenMS Proteomics Pipeline.
+
+MSGFPlusAdapter
+ Adapter for the MS-GF+ protein identification (database search) engine. More information is available [here](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_MSGFPlusAdapter.html).
+
+LuciphorAdapter
+ Adapter for the LuciPHOr2: a site localisation tool of generic post-translational modifications from tandem mass
+ spectrometry data. More information is available [here](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_LuciphorAdapter.html).
+
+pyOpenMS
+ pyOpenMS is an open-source Python library for mass spectrometry, specifically for the analysis of proteomics and
+ metabolomics data in Python. For pyOpenMS documentaion visit [this](https://pyopenms.readthedocs.io/en/latest/) link.
+
+TOPP Tools
+ All {term}`TOPP` tools can be found [here](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_documentation.html).
+
+UTILS
+ Besides TOPP, OpenMS offers a range of other tools. They are not included in TOPP as they are not part of typical
+ analysis pipelines. More information is present in [OpenMS UTILS Documentation)(https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/UTILS_documentation.html).
+
+TOPPView
+ TOPPView is a viewer for MS and HPLC-MS data. More information is available in [TOPPView documentation](topp/toppview.md).
+
+[Nightly Snapshot](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html)
+ Untested installers and containers are known as the nightly snapshot.
+
+Proteomics
+ Proteomics is the large-scale study of proteins.
+
+Proteins
+ Proteins are vital parts of living organisms, with many functions, for example composing the structural fibers of
+ muscle to the enzymes that catalyze the digestion of food to synthesizing and replicating DNA.
+
+Mascot
+ Identifies peptides in MS/MS spectra via Mascot. Please find more information in the {term}`TOPP` [Documentation](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_MascotAdapter.html).
+
+HPLC-MS
+ Data produced by High performance liquid chromatography (HPLC) separates components of a mixture, whereas mass
+ spectrometry (MS) offers the detection tools to identify them.
+
+mzML
+ The mzML format is an open, XML-based format for mass spectrometer output files, developed with the full participation
+ of vendors and researchers in order to create a single open format that would be supported by all software.
+
+mzData
+ mzData was the first attempt by the Proteomics Standards Initiative (PSI) from the Human Proteome Organization (HUPO)
+ to create a standardized format for Mass Spectrometry data.[7] This format is now deprecated, and replaced by mzML.
+
+mzXML
+ mzXML is an open data format for storage and exchange of mass spectroscopy data, developed at the SPC/Institute for
+ Systems Biology.
+
+Spectra
+ Singluar of spectrum.
+
+Spectrum
+ A mass spectrum is a type of plot of the ion signal as a function of the mass-to-charge ratio. These spectra are used
+ to determine the elemental or isotopic signature of a sample, the masses of particles and of molecules, and to
+ elucidate the chemical identity or structure of molecules and other chemical compounds.
+
+m/z
+ mass to charge ratio.
+
+RT
+ Retention time (RT).
+
+ProteoWizard
+ ProteoWizard is a set of open-source, cross-platform tools and libraries for proteomics data analyses. It provides a
+ framework for unified mass spectrometry data file access and performs standard chemistry and LCMS dataset computations.
+
+OMSSA
+ The Open Mass Spectrometry Search Algorithm (OMSSA) is an efficient search engine for identifying {term}`MS/MS`
+ {term}`peptide` {term}`spectra` by searching libraries of known protein sequences.
+
+PepNovo
+ PepNovo is a de novo sequencing algorithm for {term}`MS/MS` {term}`spectra`.
+
+De novo peptide sequencing
+ A peptide’s amino acid sequence is inferred directly from the precursor peptide mass and tandem mass spectrum
+ ({term}`MS/MS` or {term}`MS^3`) fragment ions, without comparison to a reference proteome.
+
+TOPPAS
+ An assistant for GUI-driven TOPP workflow design. It is recommended to use OpenMS through the KNIME plugins.
+```
+
diff --git a/docs/guides/contributors-quickstart-guide.md b/docs/guides/contributors-quickstart-guide.md
new file mode 100644
index 00000000..dbfb0e12
--- /dev/null
+++ b/docs/guides/contributors-quickstart-guide.md
@@ -0,0 +1,74 @@
+Contribute to OpenMS
+====================
+
+To contribute to OpenMS:
+
+- Familiarise yourself with the [OpenMS online documentation](../index.rst).
+- Learn how to [build OpenMS](../installations/build-openms-from-source.md).
+- Check out the [OpenMS tutorial for developers](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/OpenMS_tutorial.html).
+
+For any questions, please [contact us](../contact-us.md).
+
+## Technical documentation
+
+```{note}
+Untested installers and containers are known as the {term}`nightly snapshot`, are released every night. They generally pass
+automated continuous integration tests but no manual tests.
+```
+
+View the documentation for the nightly snapshot of [OpenMS develop branch](https://github.com/OpenMS/OpenMS/tree/develop)
+at the [build archive](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html).
+
+See the documentation for the [latest release](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/index.html).
+
+## Contribution guidelines
+
+Before contributing to OpenMS, read information on the development model and conventions followed to maintain a coherent
+code base.
+
+### Development model
+
+OpenMS follows the [Gitflow development workflow](http://nvie.com/posts/a-successful-git-branching-model/).
+
+Every contributor is encouraged to create their own fork (even if they are eligible to push directly to OpenMS).
+To create a fork:
+
+1. Follow the documentation on [forking](https://help.github.com/articles/fork-a-repo).
+2. Keep your fork [up-to-date](https://help.github.com/articles/syncing-a-fork).
+3. Create a [pull request](https://help.github.com/articles/using-pull-requests). Before opening the pull request, please
+ view the [pull request guidelines](../additional-resources/pull-request-checklist.md).
+
+### Coding conventions
+
+See the manual for coding style recommended by OpenMS: [Coding conventions](https://github.com/OpenMS/OpenMS/wiki/Coding-conventions).
+
+```{seealso}
+[C++ Guide](https://github.com/OpenMS/OpenMS/wiki/Cpp-Guide).
+```
+
+OpenMS automatically tests for common coding convention violations using a modified version of `cpplint`.
+Style testing can be enabled using `cmake` options. [clang-format](https://github.com/OpenMS/OpenMS/blob/develop/.clang-format) is used for formatting the cpp code.
+
+### Commit messages
+
+View the guidelines for commit messages: [How to write commit messages](https://github.com/OpenMS/OpenMS/wiki/HowTo---Write-Commit-Messages).
+
+### Automated unit tests
+
+Nightly tests run on different platforms. It is recommended to test on different platforms.
+
+```{tip}
+This saves time and increases productivity during continuous integration tests.
+```
+
+Nightly tests: [CDASH](http://cdash.openms.de/index.php?project=OpenMS).
+
+## Further contributor resources
+
+Consider the following resources for further information:
+
+- **Guidelines for adding new dependency libraries**: View the guidelines for [adding new dependency libraries](../additional-resources/developer-guidelines-for-addding-new-dependent-libraries.md).
+- **Experimental installers**: We automatically build installers for different platforms. These usually contain
+ unstable or partially untested code.
+ The nightly (unstable) installers are available at the [build archive](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/nightly/).
+- **Developer FAQ**: Visit the [Developer FAQ](../faqs/developer-faq.md) to get answers to frequently asked questions.
diff --git a/docs/guides/user-guides/user-quickstart-guide.md b/docs/guides/user-guides/user-quickstart-guide.md
new file mode 100644
index 00000000..71d07e42
--- /dev/null
+++ b/docs/guides/user-guides/user-quickstart-guide.md
@@ -0,0 +1,78 @@
+User Quickstart Guide
+====================
+
+Read the User Quickstart guide to gain a brief understanding of key concepts and how to use the tools. For more in-depth
+information, consult [OpenMS API Reference](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/index.html).
+
+## What is OpenMS
+
+[OpenMS](https://www.openms.de/) is a free, open-source C++ library with Python bindings. It is commonly used for liquid
+chromatography-mass spectrometry ({term}`LC-MS`) data management and analyses. OpenMS provides an infrastructure for the rapid
+development of mass spectrometry related software as well as a rich toolset built on top of it. OpenMS is available
+under the [three clause BSD licence](https://github.com/OpenMS/OpenMS/blob/develop/LICENSE) and runs under Windows, macOS, and Linux operating systems.
+
+## Background
+
+Before using OpenMS, become familiar with the following terms:
+
+| Tool and Utilities | Description |
+|--------------------|-------------|
+|**TOPPView** |A tool that is used to view and explore {term}`LC-MS` data, alignments, groups, peptide identifications, and more.|
+|**TOPPAS** |A graphical workflow design tool that is used to create pipelines from all {term}`TOPP tools` (and {term}`UTILS`).|
+|**TOPP tools** |A set of command line tools. Each of these command line tools is a building block of an analysis pipeline and are chained together in a way that fits the requirements of the user. The {term}`TOPP tools` are accessible from a command prompt/shell or via {term}`TOPPAS`. See also: [TOPP tutorial](../../tutorials/TOPP/TOPP-tutorial.md) and [TOPP documentation](../../topp-and-utils/topp-and-utils.md)|
+|**UTILS** |Besides {term}`TOPP`, OpenMS offers range of other tools. They are not included in {term}`TOPP` as they are not part of typical analysis pipelines. A set of command line utilities, similar to {term}`TOPP tools`, mostly used during pipeline construction or parameter optimization. |
+
+```{seealso}
+
+[UTILS documentation](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/UTILS_documentation.html)|
+```
+
+```{important}
+Users can now use {term}`KNIME` in place of {term}`TOPPAS`; the later will deprecated with no support in near future.
+Please find more information about using {term}`KNIME` in [KNIME tutorial](../../tutorials/KNIME/KNIME-tutorial.md).
+```
+
+
+## How to run a Tool
+
+A good start are the example pipelines (select **File** > **Open example file** within {term}`TOPPAS`).
+
+Read the documentation of the tools see [TOPP tutorial](../../tutorials/TOPP/TOPP-tutorial.md), [TOPP documentation](../../topp-and-utils/topp-and-utils.md) and the one of ([TOPPAS tutorial](../../tutorials/TOPPAS/TOPPAS-tutorial.md)).
+
+Alternatively, use the command line and call tools directly. In this case, you'll probably want to use some type of shell
+script for automation.
+
+## Adapt pipeline parameters
+
+The default parameters of each tool can usually be tweaked to fit the data and improve results.
+
+### Where do you change pipeline parameters?
+
+1. **TOPPAS**: Double-click the node of which you want to change the parameters of. A short docu for each parameter will
+ show up once it is selected. All parameters which would be available on the command line and in the INI
+ file are shown here as well.
+2. **Command line**: Very basic parameters can be set on the command line, e.g. `FileFilter -rt 1000:2000 .....`
+3. Doing 2 for all parameters would create a very long list, thus, use so-called `.ini` files to provide full parameter
+ sets to {term}`TOPP tools`. If no INI file is given, default parameters are used. To get a default `.ini` use
+
+ ` -write_ini `
+
+ e.g. `FileFilter -write_ini filefilter.ini`
+
+ Now, edit the INI file (which is a XML file) using the [INIFileEditor](../../topp-and-utils/ini-file-editor.md), which is another GUI tool shipped with
+ OpenMS and similar to the one build into {term}`TOPPAS`.
+
+### How do I feed the INI file to a Tool?
+
+1. **TOPPAS**: Once you changed the parameters of a node and clicked **Ok**, the parameters are in effect. Because
+ they are part of the {term}`TOPPAS` workflow, they are saved together with the workflow.
+2. **Command line** : Supply the INI file via the `-ini` flag,
+ ` -ini `
+
+ e.g. `FileFilter -ini filefilter.ini`
+
+### What parameters to set and to what value?
+
+The answer is complex, in general, read the tool description, change the parameters and compare the results using
+{term}`TOPPView` if possible. If that does not help, [contact us](../../contact-us.md). Please include all the necessary
+details we need in order to help you.
diff --git a/docs/images/additional-resources/click-fork.png b/docs/images/additional-resources/click-fork.png
new file mode 100644
index 00000000..0ca09e1e
Binary files /dev/null and b/docs/images/additional-resources/click-fork.png differ
diff --git a/docs/images/advanced-resources/Existing-Projects-into-Workspace.png b/docs/images/advanced-resources/Existing-Projects-into-Workspace.png
new file mode 100644
index 00000000..245226c5
Binary files /dev/null and b/docs/images/advanced-resources/Existing-Projects-into-Workspace.png differ
diff --git a/docs/images/advanced-resources/KNIME-Build-System.png b/docs/images/advanced-resources/KNIME-Build-System.png
new file mode 100644
index 00000000..230c84a8
Binary files /dev/null and b/docs/images/advanced-resources/KNIME-Build-System.png differ
diff --git a/docs/images/advanced-resources/KNIME-Desktop-Update-Site.png b/docs/images/advanced-resources/KNIME-Desktop-Update-Site.png
new file mode 100644
index 00000000..d1d3727e
Binary files /dev/null and b/docs/images/advanced-resources/KNIME-Desktop-Update-Site.png differ
diff --git a/docs/images/advanced-resources/KNIME-File-Handling-Nodes.png b/docs/images/advanced-resources/KNIME-File-Handling-Nodes.png
new file mode 100644
index 00000000..17b4d82d
Binary files /dev/null and b/docs/images/advanced-resources/KNIME-File-Handling-Nodes.png differ
diff --git a/docs/images/advanced-resources/eclipse-application.png b/docs/images/advanced-resources/eclipse-application.png
new file mode 100644
index 00000000..ba79e668
Binary files /dev/null and b/docs/images/advanced-resources/eclipse-application.png differ
diff --git a/docs/images/advanced-resources/new-configuration.png b/docs/images/advanced-resources/new-configuration.png
new file mode 100644
index 00000000..d8aff95c
Binary files /dev/null and b/docs/images/advanced-resources/new-configuration.png differ
diff --git a/docs/images/advanced-resources/select-root-directory.png b/docs/images/advanced-resources/select-root-directory.png
new file mode 100644
index 00000000..245226c5
Binary files /dev/null and b/docs/images/advanced-resources/select-root-directory.png differ
diff --git a/docs/images/installations/macos/copying-to-applications.png b/docs/images/installations/macos/copying-to-applications.png
new file mode 100644
index 00000000..8094de23
Binary files /dev/null and b/docs/images/installations/macos/copying-to-applications.png differ
diff --git a/docs/images/installations/macos/license-agreements.png b/docs/images/installations/macos/license-agreements.png
new file mode 100644
index 00000000..bce5463b
Binary files /dev/null and b/docs/images/installations/macos/license-agreements.png differ
diff --git a/docs/images/installations/macos/move-openms-to-applications.png b/docs/images/installations/macos/move-openms-to-applications.png
new file mode 100644
index 00000000..5647b8a2
Binary files /dev/null and b/docs/images/installations/macos/move-openms-to-applications.png differ
diff --git a/docs/images/installations/macos/opening-openms-macos.png b/docs/images/installations/macos/opening-openms-macos.png
new file mode 100644
index 00000000..1a90f732
Binary files /dev/null and b/docs/images/installations/macos/opening-openms-macos.png differ
diff --git a/docs/images/installations/macos/preparing-to-copy-to-applications.png b/docs/images/installations/macos/preparing-to-copy-to-applications.png
new file mode 100644
index 00000000..327ca3b8
Binary files /dev/null and b/docs/images/installations/macos/preparing-to-copy-to-applications.png differ
diff --git a/docs/images/installations/macos/verifying-OpenMS.png b/docs/images/installations/macos/verifying-OpenMS.png
new file mode 100644
index 00000000..6064a289
Binary files /dev/null and b/docs/images/installations/macos/verifying-OpenMS.png differ
diff --git a/docs/images/introduction/introduction_LC.png b/docs/images/introduction/introduction_LC.png
new file mode 100644
index 00000000..49151fad
Binary files /dev/null and b/docs/images/introduction/introduction_LC.png differ
diff --git a/docs/images/introduction/introduction_MS.png b/docs/images/introduction/introduction_MS.png
new file mode 100644
index 00000000..e5fd7f4e
Binary files /dev/null and b/docs/images/introduction/introduction_MS.png differ
diff --git a/docs/images/introduction/spectrum_peakmap.png b/docs/images/introduction/spectrum_peakmap.png
new file mode 100644
index 00000000..6c89ee7f
Binary files /dev/null and b/docs/images/introduction/spectrum_peakmap.png differ
diff --git a/docs/images/research/whitepapers/design-summary.png b/docs/images/research/whitepapers/design-summary.png
new file mode 100644
index 00000000..841be1c8
Binary files /dev/null and b/docs/images/research/whitepapers/design-summary.png differ
diff --git a/docs/images/research/whitepapers/summary.png b/docs/images/research/whitepapers/summary.png
new file mode 100644
index 00000000..097f5c90
Binary files /dev/null and b/docs/images/research/whitepapers/summary.png differ
diff --git a/docs/images/topp/INIFileEditor.png b/docs/images/topp/INIFileEditor.png
new file mode 100644
index 00000000..ca6e2132
Binary files /dev/null and b/docs/images/topp/INIFileEditor.png differ
diff --git a/docs/images/topp/SwathWizard.png b/docs/images/topp/SwathWizard.png
new file mode 100644
index 00000000..438f382f
Binary files /dev/null and b/docs/images/topp/SwathWizard.png differ
diff --git a/docs/images/topp/TOPPAS_simple_example.png b/docs/images/topp/TOPPAS_simple_example.png
new file mode 100644
index 00000000..d9786f9f
Binary files /dev/null and b/docs/images/topp/TOPPAS_simple_example.png differ
diff --git a/docs/images/topp/TOPPView.png b/docs/images/topp/TOPPView.png
new file mode 100644
index 00000000..85190458
Binary files /dev/null and b/docs/images/topp/TOPPView.png differ
diff --git a/docs/images/tutorials/knime/KNIME_Install.png b/docs/images/tutorials/knime/KNIME_Install.png
new file mode 100644
index 00000000..1bf43c83
Binary files /dev/null and b/docs/images/tutorials/knime/KNIME_Install.png differ
diff --git a/docs/images/tutorials/knime/KNIME_screenshot.png b/docs/images/tutorials/knime/KNIME_screenshot.png
new file mode 100644
index 00000000..f98895ca
Binary files /dev/null and b/docs/images/tutorials/knime/KNIME_screenshot.png differ
diff --git a/docs/images/tutorials/knime/KNIME_update_site.jpeg b/docs/images/tutorials/knime/KNIME_update_site.jpeg
new file mode 100644
index 00000000..cb3abbfc
Binary files /dev/null and b/docs/images/tutorials/knime/KNIME_update_site.jpeg differ
diff --git a/docs/images/tutorials/proteowizard.png b/docs/images/tutorials/proteowizard.png
new file mode 100644
index 00000000..64733b22
Binary files /dev/null and b/docs/images/tutorials/proteowizard.png differ
diff --git a/docs/images/tutorials/topp/INIFileEditor.png b/docs/images/tutorials/topp/INIFileEditor.png
new file mode 100644
index 00000000..ca6e2132
Binary files /dev/null and b/docs/images/tutorials/topp/INIFileEditor.png differ
diff --git a/docs/images/tutorials/topp/MetaDataBrowser.png b/docs/images/tutorials/topp/MetaDataBrowser.png
new file mode 100644
index 00000000..93d88edf
Binary files /dev/null and b/docs/images/tutorials/topp/MetaDataBrowser.png differ
diff --git a/docs/images/tutorials/topp/Plot2DWidget.png b/docs/images/tutorials/topp/Plot2DWidget.png
new file mode 100644
index 00000000..b9e20593
Binary files /dev/null and b/docs/images/tutorials/topp/Plot2DWidget.png differ
diff --git a/docs/images/tutorials/topp/Plot3DWidget.png b/docs/images/tutorials/topp/Plot3DWidget.png
new file mode 100644
index 00000000..4a97fafd
Binary files /dev/null and b/docs/images/tutorials/topp/Plot3DWidget.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_1D.png b/docs/images/tutorials/topp/TOPPView_1D.png
new file mode 100644
index 00000000..73e78dda
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_1D.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_help.png b/docs/images/tutorials/topp/TOPPView_help.png
new file mode 100644
index 00000000..0377524d
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_help.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_icons.png b/docs/images/tutorials/topp/TOPPView_icons.png
new file mode 100644
index 00000000..ad56c780
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_icons.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_parts.png b/docs/images/tutorials/topp/TOPPView_parts.png
new file mode 100644
index 00000000..c2507e06
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_parts.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_statistics.png b/docs/images/tutorials/topp/TOPPView_statistics.png
new file mode 100644
index 00000000..84262818
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_statistics.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools.png b/docs/images/tutorials/topp/TOPPView_tools.png
new file mode 100644
index 00000000..c7fe7cd8
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_baseline.png b/docs/images/tutorials/topp/TOPPView_tools_baseline.png
new file mode 100644
index 00000000..38b2a8cf
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_baseline.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_baseline_filtered.png b/docs/images/tutorials/topp/TOPPView_tools_baseline_filtered.png
new file mode 100644
index 00000000..8c123d46
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_baseline_filtered.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_ff_centroided.png b/docs/images/tutorials/topp/TOPPView_tools_ff_centroided.png
new file mode 100644
index 00000000..80f3b67f
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_ff_centroided.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_menu.png b/docs/images/tutorials/topp/TOPPView_tools_menu.png
new file mode 100644
index 00000000..2e70341c
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_menu.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_noisefilter.png b/docs/images/tutorials/topp/TOPPView_tools_noisefilter.png
new file mode 100644
index 00000000..56848e04
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_noisefilter.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_noisefilter_filtered.png b/docs/images/tutorials/topp/TOPPView_tools_noisefilter_filtered.png
new file mode 100644
index 00000000..eb6cc0e2
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_noisefilter_filtered.png differ
diff --git a/docs/images/tutorials/topp/TOPPView_tools_pp_picked.png b/docs/images/tutorials/topp/TOPPView_tools_pp_picked.png
new file mode 100644
index 00000000..29a7c13f
Binary files /dev/null and b/docs/images/tutorials/topp/TOPPView_tools_pp_picked.png differ
diff --git a/docs/images/tutorials/topp/TOPP_alignment.png b/docs/images/tutorials/topp/TOPP_alignment.png
new file mode 100644
index 00000000..f8bf7fb5
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_alignment.png differ
diff --git a/docs/images/tutorials/topp/TOPP_consensus_id.png b/docs/images/tutorials/topp/TOPP_consensus_id.png
new file mode 100644
index 00000000..58c5a6a2
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_consensus_id.png differ
diff --git a/docs/images/tutorials/topp/TOPP_labeled_quant.png b/docs/images/tutorials/topp/TOPP_labeled_quant.png
new file mode 100644
index 00000000..77493740
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_labeled_quant.png differ
diff --git a/docs/images/tutorials/topp/TOPP_labelfree_quant.png b/docs/images/tutorials/topp/TOPP_labelfree_quant.png
new file mode 100644
index 00000000..b24f9fb9
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_labelfree_quant.png differ
diff --git a/docs/images/tutorials/topp/TOPP_qualitycontrol.png b/docs/images/tutorials/topp/TOPP_qualitycontrol.png
new file mode 100644
index 00000000..15b0facd
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_qualitycontrol.png differ
diff --git a/docs/images/tutorials/topp/TOPP_raw_data.png b/docs/images/tutorials/topp/TOPP_raw_data.png
new file mode 100644
index 00000000..8d48a23b
Binary files /dev/null and b/docs/images/tutorials/topp/TOPP_raw_data.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_BSA_Quantitation.png b/docs/images/tutorials/toppas/TOPPAS_BSA_Quantitation.png
new file mode 100644
index 00000000..5f10bda8
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_BSA_Quantitation.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_BSA_results_2d.png b/docs/images/tutorials/toppas/TOPPAS_BSA_results_2d.png
new file mode 100644
index 00000000..1e4911e3
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_BSA_results_2d.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_BSA_results_3d.png b/docs/images/tutorials/toppas/TOPPAS_BSA_results_3d.png
new file mode 100644
index 00000000..cbf691a8
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_BSA_results_3d.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_edges.png b/docs/images/tutorials/toppas/TOPPAS_edges.png
new file mode 100644
index 00000000..02019454
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_edges.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_example_merger.png b/docs/images/tutorials/toppas/TOPPAS_example_merger.png
new file mode 100644
index 00000000..bf34aa9c
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_example_merger.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_example_profile_data_processing.png b/docs/images/tutorials/toppas/TOPPAS_example_profile_data_processing.png
new file mode 100644
index 00000000..0116c44b
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_example_profile_data_processing.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_parameters.png b/docs/images/tutorials/toppas/TOPPAS_parameters.png
new file mode 100644
index 00000000..f57a34cf
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_parameters.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_run_options.png b/docs/images/tutorials/toppas/TOPPAS_run_options.png
new file mode 100644
index 00000000..9eb57c28
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_run_options.png differ
diff --git a/docs/images/tutorials/toppas/TOPPAS_simple_example.png b/docs/images/tutorials/toppas/TOPPAS_simple_example.png
new file mode 100644
index 00000000..d9786f9f
Binary files /dev/null and b/docs/images/tutorials/toppas/TOPPAS_simple_example.png differ
diff --git a/docs/index.rst b/docs/index.rst
new file mode 100644
index 00000000..89f95730
--- /dev/null
+++ b/docs/index.rst
@@ -0,0 +1,134 @@
+About OpenMS
+============
+
+`OpenMS `_
+is an open-source software C++ library for :term:`LC-MS` data management and
+analyses. It offers an infrastructure for rapid development of mass
+spectrometry related software. OpenMS is free software available under the
+three clause BSD license and runs under Windows, macOS, and Linux.
+
+It comes with a vast variety of pre-built and ready-to-use tools for proteomics
+and metabolomics data analysis (:term:`TOPP Tools`) as well as powerful 1D, 2D and 3D
+visualization (:term:`TOPPView`).
+
+OpenMS offers analyses for various quantitation protocols, including label-free
+quantitation, :term:`SILAC`, :term:`iTRAQ`, :term:`TMT`, :term:`SRM`, :term:`SWATH`, etc.
+
+It provides built-in algorithms for de-novo identification and database search,
+as well as adapters to other state-of-the art tools like X!Tandem, :term:`Mascot`,
+OMSSA, etc. It supports easy integration of OpenMS built tools into workflow
+engines like :term:`KNIME`, Galaxy, WS-Pgrade, and :term:`TOPPAS` via the :term:`TOPP tools` concept and
+a unified parameter handling via a 'common tool description' (CTD) scheme.
+
+.. important::
+ As part of the **Center for Integrative Bioinformatics** (CiBi) in the **German Network for Bioinformatics**
+ `deNBI `_,
+ OpenMS is currently focusing the development efforts on the integration of OpenMS into KNIME. KNIME is a well-established
+ data analysis framework that supports the generation of workflows for data analysis. Using a Common Tool Description
+ (CTD) file which is writeable by every TOPP tool and a node generator program (`Generic KNIME Nodes `_), all :term:`TOPP tools` can be made available to run in KNIME.
+
+
+With :term:`pyOpenMS`, OpenMS offers Python bindings to a large part of the :term:`OpenMS API`
+to enable rapid algorithm development. OpenMS supports the Proteomics Standard
+Initiative (PSI) formats for MS data. The main contributors of OpenMS are
+currently the Eberhard-Karls-Universität in Tübingen, the Freie Universität
+Berlin, and the ETH Zürich.
+
+Contents
+--------
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Introduction
+ :titlesonly:
+
+ introduction
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Getting Started
+ :titlesonly:
+
+ installations/installation-on-gnu-linux
+ installations/installation-on-windows
+ installations/installation-on-macos
+ installations/build-openms-from-source
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Quick Start Guides
+ :titlesonly:
+
+ guides/user-guides/user-quickstart-guide
+ guides/contributors-quickstart-guide.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Tutorials
+ :titlesonly:
+
+ tutorials/TOPP/TOPP-tutorial
+ tutorials/TOPPAS/TOPPAS-tutorial
+ tutorials/KNIME/KNIME-tutorial
+
+.. toctree::
+ :maxdepth: 2
+ :caption: OpenMS TOPP Tools
+ :titlesonly:
+
+ topp-and-utils/topp-and-utils.md
+ topp-and-utils/adding-new-tool-to-topp.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Developer Resources
+ :titlesonly:
+
+ additional-resources/developer-guidelines-for-addding-new-dependent-libraries.md
+ additional-resources/external-code-using-openms.md
+ advanced-resources/custom-compilation.md
+ advanced-resources/build-custom-openms-knime-package.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: OpenMS GitHub Workflow
+ :titlesonly:
+
+ additional-resources/openms-git-workflow.md
+ additional-resources/reporting-bugs-and-issues.md
+ additional-resources/write-and-label-github-issues.md
+ additional-resources/pull-request-checklist.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Frequently Asked Questions
+ :titlesonly:
+
+ faqs/developer-faq.md
+ faqs/contributor-faq.md
+
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Downloads
+
+ downloads.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Glossary
+
+ glossary.md
+
+.. toctree::
+ :maxdepth: 2
+ :caption: Contact Us
+
+ contact-us
+
+
+Indices and tables
+==================
+
+* :ref:`genindex`
+* :ref:`search`
diff --git a/docs/installations/installation-on-gnu-linux.md b/docs/installations/installation-on-gnu-linux.md
new file mode 100644
index 00000000..60aac51b
--- /dev/null
+++ b/docs/installations/installation-on-gnu-linux.md
@@ -0,0 +1,194 @@
+Installation on GNU/Linux
+=========================
+
+## Install via Conda
+
+You can use conda to install the OpenMS library and tools without user interface. Depending on the conda channel, you can
+obtain release versions (`bioconda` channel) and nightly versions (`openms` channel).
+
+1. Follow the instructions to [install conda](https://docs.conda.io/projects/conda/en/latest/user-guide/install/linux.html).
+2. Add channels for dependencies:
+ ```bash
+ conda config --add channels defaults
+ conda config --add channels bioconda
+ conda config --add channels conda-forge
+ ```
+3. Install any of the following packages related to OpenMS
+
+```{tab} openms
+openms contains OpenMS C++ Tools.
+```
+
+```{tab} libopenms
+libopenms is the C++ library required for the OpenMS C++ Tools to work.
+```
+
+```{tab} pyopenms
+pyopenms is the python package that allows to use algorithms from libopenms in Python.
+```
+
+```{tab} openms-thirdparty
+openms-thirdparty are external tools that are wrapped in OpenMS with adapters. This is required to use the adapters in
+the openms package.
+```
+
+via `bioconda` for release versions
+
+````{tab} openms
+```bash
+conda install openms
+```
+````
+
+````{tab} libopenms
+```bash
+conda install libopenms
+```
+````
+
+````{tab} pyopenms
+```bash
+conda install pyopenms
+```
+````
+
+````{tab} openms-thirdparty
+```bash
+conda install openms-thirdparty
+```
+````
+
+or our own `openms` channel for nightly snapshots (which are build based on the same bioconda dependencies)
+
+````{tab} openms
+```bash
+conda install -c openms openms
+```
+````
+
+````{tab} libopenms
+```bash
+conda install -c openms libopenms
+```
+````
+
+````{tab} pyopenms
+```bash
+conda install -c openms pyopenms
+```
+````
+
+````{tab} openms-thirdparty
+```bash
+conda install -c openms openms-thirdparty
+```
+````
+
+## Install via Debian package
+
+For Debian-based Linux users, it is suggested to use the [deb-package](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/) provided. It is most easily installed with **[gdebi](https://launchpad.net/gdebi)**
+which automatically resolves the dependencies available in the PPA Repositories.
+
+```bash
+sudo apt-get install gdebi
+sudo gdebi /PATH/TO/OpenMS.deb
+```
+If you encounter errors with unavailable packages, troubleshoot using the following steps.
+
+1. Qt5 (or one of its packages, e.g. `qt5xbase`) is missing.
+ It might be because your Debian is too old to have a recent enough version in its official repositories. It is
+ suggested to use the same packages that are used while building (make sure to adapt the Qt version and your
+ Debian/Ubuntu version, here Xenial):
+ ```bash
+ sudo add-apt-repository ppa:beineri/opt-qt59-xenial
+ sudo apt-get update
+ ```
+ Run the installation again.
+2. ICU with its `libicu` is missing.
+ You can find the missing version on [pkgs.org](https://pkgs.org) and install it with `gdebi`, too. You can have
+ multiple versions of ICU installed.
+3. Error while executing a tool
+ To ensure the tool functionality, make sure you add the `OPENMS_DATA_PATH` variable to your environment as follow
+ `export OPENMS_DATA_PATH=/usr/share/OpenMS`
+4. Thirdparty installation of Qt5 in 1
+ Make sure you source the provided environment file using:
+ `source /opt/qt59/bin/qt59-env.sh`
+
+ Executables for THIRDPARTY applications can be found in:
+ `/usr/share/OpenMS/THIRDPARTY`
+5. Add the folders in your `PATH` for a convenient use of the adapters.
+
+## Install via package managers
+
+Packaged versions of **OpenMS** are provided for Fedora, OpenSUSE, Debian, and Ubuntu. You can find them to download
+[here](https://pkgs.org/download/openms). For other GNU/Linux distributions or to obtain the most recent version of the
+library, installation should be done via building from the source code.
+
+```{important}
+These packages are not directly maintained by the OpenMS team and they can not be guaranteed to have the
+same behaviour as when building it from source code. Also, their availability and version is subject to change and
+support might be limited (due to unforeseen or untested behaviour). It is suggested not to install them parallel to our
+Debian package.
+```
+
+```{note}
+Some thirdparty software used via adapter tools in OpenMS might also require an installed JavaVM.
+```
+
+## Run via a (Bio)Container
+
+Install a containerization software (e.g., [Docker](https://docs.docker.com/engine/install/) or [Singularity](https://sylabs.io/guides/3.0/user-guide/quick_start.html#quick-installation-steps))
+
+Our container support is constantly updated. Docker images provided by us,
+can be obtained via [ghcr.io](https://ghcr.io).
+
+1. [openms-executables](https://ghcr.io/openms/openms-executables)
+2. [openms-library](https://ghcr.io/openms/openms-library)
+
+Otherwise, the [BioContainers Registries](https://biocontainers.pro/registry) and the associated Galaxy
+project provide native containers from our bioconda packages for both Docker and Singularity.
+
+1. [BioContainers libopenms](https://biocontainers.pro/tools/libopenms)
+2. [BioContainers openms](https://biocontainers.pro/tools/openms)
+3. [BioContainers openms-thirdparty](https://biocontainers.pro/tools/openms-thirdparty)
+4. [BioContainers pyOpenMS](https://biocontainers.pro/tools/pyopenms)
+
+Images of the containers can be pulled via or one of the following commands:
+
+````{tab} Docker
+```bash
+docker pull quay.io/biocontainers/libopenms
+docker pull quay.io/biocontainers/openms
+docker pull quay.io/biocontainers/pyopenms
+docker pull quay.io/biocontainers/openms-thirdparty
+```
+````
+
+````{tab} Singularity
+```bash
+docker pull https://depot.galaxyproject.org/singularity/libopenms
+docker pull https://depot.galaxyproject.org/singularity/openms
+docker pull https://depot.galaxyproject.org/singularity/pyopenms
+docker pull https://depot.galaxyproject.org/singularity/openms-thirdparty
+```
+````
+
+If Singularity images fail to download or run, try to use the Docker images as Singularity will automatically convert them.
+
+Docker images from our own continuous integration can be installed via the following commands:
+
+```bash
+docker pull ghcr.io/openms/openms-library
+docker pull ghcr.io/openms/openms-executables
+```
+
+per default this results in the download of the latest nightly snapshot. Specify a release version to
+receive a stable version.
+
+Dockerfiles to build different kind of images (corresponding to build instructions, e.g. on ArchLinux) yourself can be found on
+GitHub in our [OpenMS/dockerfiles](https://github.com/OpenMS/dockerfiles) repository.
+
+## Build OpenMS from source
+
+To build OpenMS from source, follow the build instructions for [Linux](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/install_linux.html).
+
diff --git a/docs/installations/installation-on-macos.md b/docs/installations/installation-on-macos.md
new file mode 100644
index 00000000..48b5c61e
--- /dev/null
+++ b/docs/installations/installation-on-macos.md
@@ -0,0 +1,92 @@
+Installation on macOS
+====================
+
+## Install via macOS installer
+
+To install OpenMS on macOS, run the following steps:
+
+1. Download and install the macOS drag-and-drop installer from the [archive](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/).
+2. Double click on the dowloaded file. It will start to open the download `openms--macos.dmg` file
+
+
+
+3. Verify the download.
+
+
+
+4. Agree the license agreements.
+
+
+
+5. Drag openms to applications.
+
+
+
+6. It will start copying to applications.
+
+
+
+
+
+To use {term}`TOPP` as regular app in the shell, add the following lines to the `~/.profile` file.
+
+```bash
+export OPENMS_TOPP_PATH=
+source ${OPENMS_TOPP_PATH}/.TOPP_bash_profile
+```
+
+Make sure `` points to the folder where OpenMS is installed locally (e.g., `/Applications/OpenMS-`)
+
+## Install via Conda with the Bioconda channel
+
+Follow the instructions for {ref}`GNU/Linux <./installation-on-gnu-linux.md#install-via-conda>`. Use the Terminal.app or any
+terminal of your choice to run the commands.
+
+```{note}
+Due to unavailability of an exact copy of the macOS build machines used in bioconda, we currently cannot offer nightly
+builds for macOS on our own conda channel `openms`.
+```
+
+## Run via (Bio)Container
+
+Follow the instructions for{ref}`GNU/Linux <./installation-on-gnu-linux.md#run-via-a-bio-container>`. Make sure to
+follow specific installation instructions for macOS during the installation of the containerization software.
+Use the Terminal.app or any terminal of your choice to run the commands.
+
+```{note}
+Running OpenMS containers (currently built for Linux) on macOS involves
+running virtual machines (sometimes hidden from the user, e.g., with Docker).
+This comes with disadvantages in runtime and memory consumption.
+```
+
+
+## Build OpenMS from source
+
+To build OpenMS from source, follow the build instructions for [macOS](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/install_mac.html).
+
+## Known Issues
+
+1. OpenMS software landing in quarantine since macOS Catalina after installation of the `.dmg`.
+
+ Since macOS Catalina (maybe also Mojave) notarized apps and executables are mandatory.
+
+ ```{important}
+ Although there is a lot of effort in signing and notarizing everything, it seems like openms software
+ still lands in quarantine on the above mentioned systems, after installation of the DMG (when downloading it from a
+ browser).
+ ```
+
+ To have a streamlined experience without blocking popups, it is recommended to remove the quarantine flag manually,
+ using the following steps:
+
+ Open the Terminal.app and type the following (replace the first line with the actual installation directory):
+ ```bash
+ cd /Applications/OpenMS-
+ sudo xattr -r -d com.apple.quarantine *
+ ```
+2. Bug with running Java based thirdparty tools like {term}`MSGFPlusAdapter` and {term}`LuciphorAdapter` from within **TOPPAS.app**
+
+ If you face issues while running Java based thirdparty tools from within {term}`TOPPAS.app`, run the {term}`TOPPAS.app`
+ from within the Terminal.app (e.g. with the `open` command) to get access to the path where Java is located.
+ Java is usually present in the `PATH` of the terminal. Advanced users can set this path in the `Info.plist` of/inside
+ the TOPPAS.app.
diff --git a/docs/installations/installation-on-windows.md b/docs/installations/installation-on-windows.md
new file mode 100644
index 00000000..9ba0b18a
--- /dev/null
+++ b/docs/installations/installation-on-windows.md
@@ -0,0 +1,46 @@
+Installation on Windows
+=======================
+
+## Install via Windows installer
+
+To Install the binary package of OpenMS & {term}`TOPP`:
+
+1. Download and execute the installer `OpenMS--Win64.exe` from the [archive](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/) and follow its instructions.
+2. Run the installer under the user account that later runs OpenMS. It is not advised to use admin account for
+ installation. When asked for an admin authentication, please enter the credentials.
+
+```{tip}
+The windows binary version works with most versions of windows from Win7 to Win10 (older versions might
+ still work but are untested).
+```
+
+## Build OpenMS from source
+
+To build OpenMS from source, follow the build instructions for [Windows](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/install_win.html).
+
+## Known issues
+
+1. During installation, an error message pops up, saying "The installation of the Microsoft .NET 3.5 SP1' package failed!
+
+ You must download and install it manually in order for Proteowizard to work.
+ This should only happen if installion is done by selecting the "Third Party - Proteowizard" components. The reason is
+ usually that **.NET 3.5 SP1** is already installed (see Windows Control Panel). If it's not installed, follow the
+ instructions of the error message.
+2. During installation, an error message pops up, saying:
+ > "The installation of the Visual Studio redistributable package ... failed. ..."
+
+ This is a known issue with a Microsoft package, we cannot do anything about it.
+ The error message will give the location where the redistributable package was extracted to. Go to this folder and
+ run the executable (usually named `vcredistXXXX.exe`) as an administrator (right-click and then select **Run-As**). You will likely
+ receive an error message (this is also the reason why the OpenMS setup complained about it). You might have to find
+ the solution to fix the problem in your local machine. If you're lucky the error message is instructive and the
+ problem is easy to fix.
+3. Error: "Error opening installation log file"
+
+ To fix, check the system environment variables. Make sure they are apt. There should a `TMP` and a `TEMP` variable,
+ and both should contain one directory only, which exists and is writable. Fix accordingly (search the internet on
+ how to change environment variables on Windows).
+4. For Win8 or later, Windows will report an error while installing `.net4` as it's mostly included. But it might occur
+ that `.net3.5` does not get properly installed during the process.
+
+ Fix is to enable the .NET Framework 3.5 yourself through Control Panel. See this [Microsoft help page](https://msdn.microsoft.com/de-de/library/hh506443(v=vs.110).aspx#ControlPanel) for detailed information. Even if this step fails, this does not affect the functionality of OpenMS, except for the executability of included thirdparty tools (ProteoWizard).
diff --git a/docs/introduction.md b/docs/introduction.md
new file mode 100644
index 00000000..c8d6ef45
--- /dev/null
+++ b/docs/introduction.md
@@ -0,0 +1,119 @@
+Introduction
+============
+
+[OpenMS](http://www.openms.org/)
+is an open-source software C++ library for {term}`LC-MS` data management and
+analyses. It offers an infrastructure for rapid development of mass
+spectrometry related software. OpenMS is free software available under the
+three clause BSD license and runs under Windows, macOS, and Linux.
+
+```{note}
+This introduction is aimed at users new to the field of LC-MS data analysis and will introduce some basics terms
+and concepts. How to handle the data analysis, available data structures, algorithms and more are covered in the various
+subsections of this documentation.
+```
+
+# Background
+
+Proteomics and metabolomics are interdisciplinary research fields that study structure, function, and interaction of
+proteins and metabolites. They employ large-scale experimental techniques that allow acquiring data at the level of
+cellular systems to whole organisms. Mass spectrometry combined with chromatographic separation is commonly used to
+identify, characterize or quantify the amount of proteins and metabolites.
+
+In mass spectrometry-based proteomics and metabolomics, biological samples are extracted, prepared, and separated to
+reduce sample complexity. The separated analytes are ionized and measured in the mass spectrometer. Mass and abundance
+of ions are stored in mass spectra and used to identify and quantify the analytes in the sample using computational
+methods. The quantity and identity of analytes can then be used, for instance, in biomarker discovery, medical diagnostics,
+or basic research.
+
+## Liquid Chromatography(LC)
+
+LC aims to reduce the complexity of the measured sample by separating analytes based on their physicochemical properties.
+Separating analytes in time ensures that a manageable amount of analytes elute at the same time. In mass
+spectrometry-based proteomics, (high-pressure) liquid chromatographic separation techniques (HPLC) are methods of choice
+to achieve a high degree of separation. In HPLC, {term}`peptides` are separated on a column. Solved in a pressurized liquid
+(mobile phase) they are pumped through a solid adsorbent material (stationary phase) packet into a capillary column.
+Physicochemical properties of each peptide determine how strongly it interacts with the stationary phase. The most
+common HPLC technique in proteomics and metabolomics uses reversed-phase chromatography (RPC) columns. RPC employs a
+hydrophobic stationary phase like {term}`octadecyl (C18)`, a nonpolar carbon chain bonded to a silica base, and a polar mobile
+phase. Polar molecules interact weakly with the stationary phase and elute earlier, while non-polar molecules are retained.
+Interaction can be further modulated by changing the gradient of solvent concentration in the mobile phase over time.
+Elution times in LC are inherently prone to variation, for example, due to fluctuations in the flow rate of the mobile
+phase or change of column. Retention time shifts between runs may be compensated using computational chromatographic
+retention time alignment methods. In the {term}`LC-MS` setup, the column is directly coupled to the ion source of the mass
+spectrometer.
+
+
+
+## Mass Spectrometry
+
+MS is an analytical technique used to determine the {term}`mass` of molecules. In order to achieve highly accurate and sensitive
+mass measurements at the atomic scale, mass spectrometers manipulate charged particles using magnetic and electrostatic
+fields.
+
+
+
+In a typical mass spectrometer, three principal components can be identified:
+
+- **Ion Source**: A mass spectrometer only handles {term}`ions`. Thus, charge needs first be transferred to uncharged
+ particles. The component responsible for the ionization is the ion source. Different types of ion sources and ionization
+ techniques exist with {term}`electrospray ionization (ESI)` being currently the most widely used ionization technique
+ for mass spectrometry-based proteomics.
+
+- **Mass Analyzer**: Most commonly used mass analyzer in proteomics are {term}`time-of-flight (TOF)` mass analyzers,
+ {term}`quadrupole mass filters`, and {term}`orbitrap analyzers`. In TOF mass analyzers, the ions are accelerated in an electric field.
+ The flight time of an ion allows calculating the velocity which in turn is used to calculate the mass-to-charge ratio
+ (m/z). Varying the electric field allows filtering certain mass-to-charge ratios before they enter the detector. In
+ quadrupole mass filters, ions pass through an oscillating electric field created by four parallel rods. For a
+ particular voltage, only ions in a certain mass-to-charge range will reach the detector. The orbitrap is an ion trap
+ mass analyzer (and detector) that traps ions in orbital motion between a barrel-like outer electrode and a spindle-like
+ central electrode allowing for prolonged mass measurement. As a result of the prolonged mass measurements, a high mass
+ resolution can be achieved.
+
+- **Detector**: The last component of the mass spectrometer is the detector. It determines the abundance of ions that
+ passed through the mass analyzer. Ion intensities (a value that relates to its abundance) and the mass-to-charge ratio
+ are recorded in a mass spectrum.
+
+A sample is measured over the retention time of the chromatography typically resulting in tens of thousands of spectra.
+The measurement of one sample is called an MS run and the set of spectra called an {term}`MS(1)` map or peak map.
+
+
+
+The left image displays spectrum with peaks (m/z and intensity values) and the right image shows spectra stacked in
+retention time yielding a peak map.
+
+In proteomics and metabolomics, the {term}`MS(1)` intensity is often used for the quantification of an analyte. Identification
+based on the {term}`MS(1)` mass-to-charge and the isotope pattern is highly ambiguous. To improve identification, tandem mass
+spectrometry {term}`(MS/MS)` can be applied to assess the analyte substructure. To this end, the precursor ion is isolated and
+kinetically fragmented using an inert gas (e.g., Argon). Fragments produced by {term}`collision-induced dissociation (CID)`
+are stored in an {term}`MS^2` (or {term}`MS/MS`) spectrum and provide information that helps to resolve the ambiguities in identification.
+Alternatively, {term}`MS/MS` spectra can be used for quantification.
+
+Get started with installing OpenMS using the installers available for different operating systems!
+
+## Installation on different platforms
+
+```{tab} GNU/Linux
+
+```bash
+wget https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/OpenMS-2.8.0-Debian-Linux-x86_64.deb
+
+```
+
+```
+
+```{tab} Windows
+
+```bash
+wget https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/OpenMS-2.8.0-Win64.exe
+```
+
+```
+
+```{tab} macOS
+
+```bash
+wget https://abibuilder.informatik.uni-tuebingen.de/archive/openms/OpenMSInstaller/release/latest/OpenMS-2.8.0-macOS.dmg
+```
+
+```
diff --git a/docs/research/whitepapers/OpenMS-2.0-kernel-whitepaper.md b/docs/research/whitepapers/OpenMS-2.0-kernel-whitepaper.md
new file mode 100644
index 00000000..719e571c
--- /dev/null
+++ b/docs/research/whitepapers/OpenMS-2.0-kernel-whitepaper.md
@@ -0,0 +1,122 @@
+OpenMS 2.0 Kernel Whitepaper
+===========================
+
+## Introduction
+
+With the constantly increasing amount of data that needs to be handled in computational mass spectrometry we start to
+reach the boundary of what is possible given the current design and implementation of the OpenMS kernel. Already in 2012
+the OpenMS core team discussed and identified the need for a redesign of the OpenMS kernel with respect to it's scalability
+and ease of use. So far, only partial solutions have been proposed that lack the approval of the OpenMS core team as
+well as the OpenMS community.
+
+This document summarises the discussion of the OpenMS core team, held at the OpenMS developer retreat in Izmir,
+Turkey 2014.
+
+## Detailed problem description
+
+With modern mass spectrometry instruments and measurement techniques the amounts of data that need to be analysed in
+computational mass spectrometry reach magnitudes that do not fit into the main memory of neither desktop nor clusters
+machines. Therefore the OpenMS kernel has to be redesigned to decouple the algorithms from the actual implementation of
+the data structures.
+
+Here we describe the interfaces and concepts we plan to implement for OpenMS 2.0. The OpenMS team has acknowledged the
+fact, that this will only be possible in an incremental way, i.e., implementing those interfaces on top of the existing
+data structures and then switch the individual classes to the new design, instead of a large redesign in a separate
+development line.
+
+## Benchmarking the changes
+
+Before we start to implement individual changes we intend to create a benchmark suite. This benchmark suite should
+enable us to assess the changes in memory consumption and runtime due to the new data structures.
+
+## Spectrum/Chromatogram interface
+
+The first decision reached during the discussion was to abandon the `Peak` data structure. It will be replaced by a pair
+of `double`s such that a spectrum/chromatogram is a combination of two `std::vector`.
+
+```cpp
+class RawDataArray
+{
+ std::vector intensities_;
+ std::vector positions_;
+};
+```
+
+with invariant `intensities.size() == positions.size()`. This data structure will be augmented with a container to hold
+position-wise meta data (see for instance `FloatDataArray`).
+
+```cpp
+class RawDataArray
+{
+ typedef std::vector MetaDataArray;
+ typedef std::string MetaDataID;
+
+ std::vector intensities_;
+ std::vector positions_;
+ std::map meta_;
+};
+```
+
+Given this base class `Spectrum` and `Chromatogram` can be derived from this class, redefining `position_` to either
+`m/z` or `rt`. Additionally the `Spectrum` and `Chromatogram` will add relevant members (e.g., MS-level for `Spectrum`)
+that should not be stored as meta data.
+
+```
+class Spectrum : public RawDataArray
+{
+ int ms_level_;
+ Precursor precursor_;
+ double rt_;
+ int scan_id_;
+};
+```
+
+The overall design is summarised in the following figure.
+
+
+
+## MSRun (was MSExperiment)
+
+`MSExperiment` will be renamed to `MSRun`. On top of MSRun we will define an interface that hides the actual implementation
+of `MSRun` from the user allowing the use of cached, on-disc, or full in-memory implementations. Direct access to
+chromatograms will be hidden behind the `ISpectrumAccess` and `IChromatogramAccess` interfaces.
+
+```cpp
+class ISpectrumAccess
+{
+public:
+ int getNrSpectra() const;
+ getMetaSpectrum(int i) const;
+ const ISpectrum& getSpectrum(int i) const;
+ const ExpMeta& getExperimentMeta() const;
+ // iterators for spectra
+ SpectrumIterator spectrumBegin();
+ SpectrumIterator spectrumEnd();
+};
+
+class IChromatogramAccess
+{
+public:
+ int getNrChromatograms() const;
+ getMetaSpectrum(int i) const;
+ const IChromatogram& getChromatogram(int i) const;
+ const ExpMeta& getExperimentMeta() const;
+ // iterators for chromatograms
+ ChromatogramIterator chromatogramBegin();
+ ChromatogramIterator chromatogramEnd();
+};
+```
+
+The overall design is summarised in the following figure.
+
+
+
+## Modularisation
+
+We decided that the OpenMS library has to be modularised. Starting with SuperHirn as a proof of concept we will identify
+other parts of the library that should be an independent module and move them into a separate library. Additionally we
+will add a new library `libOpenMSChemistry` that should implement light-weight alternatives to the existing chemistry
+data structures.
+
+To avoid the direct integration of classes associated to UTILS into OpenMS, we will additionally support multi-file UTILS
+compared to the existing TOPP like utils, that should consist of only one file.
diff --git a/docs/topp-and-utils/adding-new-tool-to-topp.md b/docs/topp-and-utils/adding-new-tool-to-topp.md
new file mode 100644
index 00000000..36b9082c
--- /dev/null
+++ b/docs/topp-and-utils/adding-new-tool-to-topp.md
@@ -0,0 +1,100 @@
+Adding New Tool to The TOPP suite
+=====================================
+
+## The OpenMS pipeline (TOPP)
+
+Any tool that is written with the OpenMS library can easily be made into a TOPP tool by simply using the OpenMS command
+line parser which is able to parse ParamXML, a powerful XML based description of the tool. Hence most analysis algorithms
+in OpenMS are available as a stand-alone tool which can be called on the command line or integrated into workflow engines
+via the CTD mechanism. A current list of TOPP tools can be found in [the documentation](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/release/latest/html/TOPP_documentation.html).
+
+## What do I have to do to add a new TOPP tool?
+
+The recommended way is to inherit from the class TOPPBase as in existing TOPP tools (sources available in /src/topp/). This will add command line parsing functionality to your tool as described in the TOPP section of this page.
+
+- Add the code to `src/topp/` and register it in `src/topp/executables.cmake`
+- Add your tool (with the correct category) to `getTOPPToolList()` in `src/openms/source/APPLICATIONS/ToolHandler.cpp`.
+ This creates a doxygen page with the `–help` output of the tool (using `TOPPDocumenter`). This page must be included
+ at the end of the doxygen documentation of your tool (see other tools for an example).
+- Add it to the TOPP docu page (in `doc/doxygen/public/TOPP.doxygen`)
+- Add the name to `src/topp/executables.cmake`
+- Write a TOPP test (add it to `src/tests/topp/CMakeLists.txt`)
+
+```{warning}
+Handle any kind of input files to your TOPP tool via command line flags and use the `${DATA_DIR_TOPP}` prefix. Use
+ini-files to specify output-files, but not input-files. Doing otherwise will break out-of-source builds.
+```
+
+```{hint}
+add `-test` to the call of your TOPP tool and also create the expected output that you put in `src/tests/topp` with that
+flag active. The flag ensures that UniqueId's, dates etc are equal no matter where and when the tool is run.
+```
+
+## What do I have to do to add a new UTILS tool?
+
+- Add the code to `src/utils/` and register it in `src/utils/executables.cmake`.
+- Add your tool to `getUtilList()` in `src/openms/source/APPLICATIONS/ToolHandler.cpp`. This creates a doxygen page with
+ the `–help` output of the tool (using `TOPPDocumenter`). This page must be included at the end of the doxygen
+ documentation of your tool (see other tools for an example).
+- Add it to the UTILS docu page (in `doc/doxygen/public/UTILS.doxygen`)
+- Write a test (this is optional for UTILS). See TOPP tools above and add the test to the bottom of `src/tests/topp/CMakeLists.txt`.
+
+## I want to implement a new file adapter. What is to be done?
+
+First, add a file adapter class to the `include/OpenMS/FORMAT/` and `source/FORMAT/` folders. The file adapter should
+implement a default constructor, a load method and a store method. Make sure your code conforms to the OpenMS Coding
+conventions. For automatic file type recognition, you need to
+
+- register your new file type at the Type enum in `/include/OpenMS/FORMAT/FileTypes.h`,
+- flag the file type as supported in the isSupported method of `/source/FORMAT/FileHandler.C`
+- register the file extension in the getTypeByFileName method of `/source/FORMAT/FileHandler.C`
+
+If the new file is a peak or feature file format you should also add it to loadExperiment or loadFeatures, respectively,
+of the FileHandler class. To add the file format to the TOPPView open dialog, you have to modify the file
+`/source/APPLICATIONS/TOPPViewBase.C`.
+
+- Add the file extensions to the filter_all and filter_single variables of the getFileList_ method.
+
+To add your format to TOPP applications:
+
+- add the file extension to the extensions list of the respective parameter:
+ ```
+ e.g. setValidStrings_("in_type", StringList::create("mzData,mzXML,mzML")); in FileInfo
+ ```
+
+## How to create an icon file for a TOPP tool under Windows?
+
+- Create an .ico file: first, you need some graphics program (The GIMP is recommended) think of a motive and remind
+ yourself that you have limited space. Create at least a 16x16, 32x32, 48x48 and 64x64 pixel version and save each of
+ them in a separate layer of the respective size. Do not add any larger sized layers, since Win XP will not display any
+ icon then. When saving the image as type `.ico` the GIMP will ask you for the color depth of each layer. As it is
+ recommended to have multiple color depths of each icon-size, go back to the layers and duplicate each layer twice.
+ That should give you 12 layers. Now, save the image as `.ico` (e.g. TOPPView.ico) file, giving each group of equal
+ sized layers a 32 bit (8 bit transparency), 8 bit (1 bit transparency), 4 bit (1 bit transparency) color depth.
+
+```{attention}
+Make sure to assign the higher color depth to the upper layers as Windows will not pick the highest possible color
+otherwise.
+```
+
+- Create a resource file: Create a text file named `.rc` (e.g. TOPPView.rc) Insert the following line: 101 ICON
+ "TOPPView.ico" , replacing TOPPView with your binary name. Put both files in `OpenMS/source/APPLICATIONS/TOPP/`
+ (similar files for other TOPP tools already present). Re-run cmake and re-link your TOPP tool.
+
+Voila. You should have an iconized TOPP tool.
+
+## Develop your Tool in an external project using OpenMS
+
+To include the OpenMS library in one of your projects, we recommend to have a look at a small emulated external project
+in our repository. We strongly suggest to use CMake for building your project together with OpenMS to make use of the
+macros and environment information generated during the build of the OpenMS library.
+
+## The Common Tool Description (CTD)
+
+The CTD is a format developed from the OpenMS team to allow the user to use TOPP tools also in other workflow engines.
+Each tool can output a CTD description of itself (the XML scheme for the CTD can be found here), which can then be used
+by a node generator program to generate nodes for different workflow engines. The CTD mechanism is shared by OpenMS with
+other mature libraries like SeqAn and BALL. An example for a node generation program are the Generic KNIME Nodes. The
+most complete description on how to generate your own Generic KNIME Nodes based on a CTD (e.g. from your freshly
+developed command line tool), can be found on the SeqAn documentation. We are working on a tutorial specifically
+tailored to OpenMS.
diff --git a/docs/topp-and-utils/ini-file-editor.md b/docs/topp-and-utils/ini-file-editor.md
new file mode 100644
index 00000000..541316c1
--- /dev/null
+++ b/docs/topp-and-utils/ini-file-editor.md
@@ -0,0 +1,10 @@
+INIFileEditor
+============
+
+Can be used to visually edit INI files of TOPP tools.
+
+The values can be edited by double-clicking or pressing F2.
+
+The documentation of each value is shown in the text area on the bottom of the widget.
+
+
diff --git a/docs/topp-and-utils/proteomicslfq.md b/docs/topp-and-utils/proteomicslfq.md
new file mode 100644
index 00000000..55bd1122
--- /dev/null
+++ b/docs/topp-and-utils/proteomicslfq.md
@@ -0,0 +1,49 @@
+ProteomicsLFQ
+=============
+
+
+ProteomicsLFQ performs label-free quantification of peptides and proteins.
+
+## Input
+
+- Spectra in mzML format
+- Identifications in idXML or mzIdentML format with posterior error probabilities as score type. To generate those we
+ suggest to run:
+ - PeptideIndexer to annotate target and decoy information.
+ - PSMFeatureExtractor to annotate percolator features.
+ - PercolatorAdapter tool (score_type = 'q-value', -post-processing-tdc).
+ - IDFilter (pep:score = 0.01) to filter PSMs at 1% FDR.
+- An experimental design file:
+
+ ```{seealso}
+ [Experimental Design](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/classOpenMS_1_1ExperimentalDesign.html) for details.
+ ```
+- A protein database in with appended decoy sequences in FASTA format.
+ (e.g., generated by the [OpenMS DecoyDatabase tool](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/namespaceOpenMS.html)).
+ Processing:
+ ProteomicsLFQ has different methods to extract features: ID-based (targeted only), or both ID-based and untargeted.
+ - The first method uses targeted feature dectection using RT and m/z information derived from identification data to
+ extract features.
+ ```{note}
+ Only identifications found in a particular MS run are used to extract features in the same run. No transfer of IDs
+ (match between runs) is performed.
+ ```
+ - The second method adds untargeted feature detection to obtain quantities from unidentified features. Transfer of Ids
+ (match between runs) is performed by transfering feature identifications to coeluting, unidentified features with
+ similar mass and RT in other runs.
+
+## Requantification
+
+Optionally, a requantification step is performed that tries to fill NA values. If a peptide has been quantified in
+more than half of all maps, the peptide is selected for requantification. In that case, the mean observed RT
+(and theoretical m/z) of the peptide is used to perform a second round of targeted extraction.
+
+## Output
+
+Output format are as follows:
+
+- mzTab file with analysis results.
+- MSstats file with analysis results for statistical downstream analysis in MSstats.
+- ConsensusXML file for visualization and further processing in OpenMS.
+
+Potential scripts to perform the search can be found under `src/tests/topp/ProteomicsLFQTestScripts`.
diff --git a/docs/topp-and-utils/swathwizard.md b/docs/topp-and-utils/swathwizard.md
new file mode 100644
index 00000000..22412fdd
--- /dev/null
+++ b/docs/topp-and-utils/swathwizard.md
@@ -0,0 +1,24 @@
+SwathWizard
+==========
+
+An assistant for Swath analysis.
+
+The Wizard takes the user through the whole analysis pipeline for SWATH proteomics data analysis, i.e. the
+[TOPP Documentation: OpenSwathWorkflow](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/UTILS_OpenSwathWorkflow.html) tool, including downstream tools such as [GitHub:PyProphet/pyProphet](https://github.com/PyProphet/pyprophet) and the [GitHub:msproteomicstools/TRIC alignment](https://github.com/msproteomicstools/msproteomicstools) tool.
+
+Since the downstream tools require Python and the respective modules, the Wizard will check their proper installation
+status and warn the user if a component is missing.
+
+Users can enter the required input data (mzML MS/MS data, configuration files) in dedicated fields, usually by drag and
+droping files from the operating systems' file explorer (Explorer, Nautilus, Finder...). The output of the Wizard is
+both the intermediate files from OpenSWATH (e.g. the XIC data in `.sqMass` format) and the tab-separated table format
+(`.tsv`) from pyProphet and TRIC.
+
+This is how the wizard looks like:
+
+
+
+A schematic of the internal data flow (all tools are called by SwathWizard in the background) can be found in the
+[TOPP Documentation: SwathWizard](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_SwathWizard.html).
+
+A recommended test data for the Wizard is the [PASS00779](https://db.systemsbiology.net/sbeams/cgi/PeptideAtlas/PASS_View?identifier=PASS00779) dataset.
diff --git a/docs/topp-and-utils/topp-and-utils.md b/docs/topp-and-utils/topp-and-utils.md
new file mode 100644
index 00000000..23f0d78e
--- /dev/null
+++ b/docs/topp-and-utils/topp-and-utils.md
@@ -0,0 +1,44 @@
+TOPP
+====
+
+**TOPP - The OpenMS Proteomics Pipeline** is a pipeline for the analysis of HPLC-MS data. It consists of several small
+applications that can be chained to create analysis pipelines tailored for a specific problem.
+
+The TOPP tools are divided into several subgroups, like graphical tools, file handling, signal processing and
+preprocessing, quantitation, map alignment, protein/peptide identification, protein/peptide processing, targeted
+experiments and OpenSWATH, peptide property prediction, cross-linking, quality-control, among a few.
+
+Few of the graphical tools are explained below:
+
+```{toctree}
+:maxdepth: 1
+
+toppview
+toppas
+ini-file-editor
+swathwizard
+```
+
+For advanced documentation on every TOPP tool, see the [OpenMS TOPP API Reference](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_documentation.html).
+
+UTILS
+=====
+
+Besides {term}`TOPP`, OpenMS offers range of other tools. They are not included in {term}`TOPP` as they are not part of
+typical analysis pipelines. A set of command line utilities, similar to {term}`TOPP tools`, mostly used during pipeline
+construction or parameter optimization.
+
+
+```{seealso}
+
+[UTILS documentation](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/UTILS_documentation.html)
+
+```
+
+Let's explore one such utitliy tool:
+
+```{toctree}
+:maxdepth: 1
+
+proteomicslfq
+```
diff --git a/docs/topp-and-utils/toppas.md b/docs/topp-and-utils/toppas.md
new file mode 100644
index 00000000..cd496144
--- /dev/null
+++ b/docs/topp-and-utils/toppas.md
@@ -0,0 +1,30 @@
+TOPPAS
+======
+
+An assistant for GUI-driven TOPP workflow design.
+
+**TOPPAS** allows to create, edit, open, save, and run TOPP workflows. Pipelines can be created conveniently in a GUI by
+means of mouse interactions. The parameters of all involved tools can be edited within the application and are also
+saved as part of the pipeline definition in the `.toppas` file. Furthermore, TOPPAS interactively performs validity
+checks during the pipeline editing process, in order to make it more difficult to create an invalid workflow. Once set
+up and saved, a workflow can also be run without the GUI using the `ExecutePipeline` TOPP tool.
+
+The following figure shows a simple example pipeline that has just been created and executed successfully:
+
+
+
+More information about TOPPAS can be found in the [TOPPAS tutorial](../tutorials/TOPP/TOPPAS-tutorial.md).
+
+**The command line parameters of this tool are**:
+
+```bash
+TOPPAS -- An assistant for GUI-driven TOPP workflow design.
+
+Usage:
+ TOPPAS [options] [files]
+
+Options are:
+ --help Shows this help
+ --debug Enables debug messages
+ -ini Sets the INI file (default: ~/.TOPPAS.ini)
+```
diff --git a/docs/topp-and-utils/toppview.md b/docs/topp-and-utils/toppview.md
new file mode 100644
index 00000000..0b001e51
--- /dev/null
+++ b/docs/topp-and-utils/toppview.md
@@ -0,0 +1,34 @@
+TOPPView
+=======
+
+**TOPPView** is a viewer for MS and HPLC-MS data. It can be used to inspect files in mzML, mzData, mzXML and several other
+file formats. It also supports viewing data from an OpenMS database. The following figure shows two instances of TOPPView
+displaying a HPLC-MS map and a MS raw spectrum:
+
+
+
+More information about TOPPView can be found in the [TOPP tutorial](../tutorials/TOPP/TOPP-tutorial.md).
+
+**The command line parameters of this tool are**:
+
+```bash
+TOPPView -- A viewer for mass spectrometry data.
+
+Usage:
+ TOPPView [options] [files]
+
+Options are:
+ --help Shows this help
+ -ini Sets the INI file (default: ~/.TOPPView.ini)
+ --force Forces scan for new tools/utils
+
+Hints:
+ - To open several files in one window put a '+' in between the files.
+ - '@bw' after a map file displays the dots in a white to black gradient.
+ - '@bg' after a map file displays the dots in a grey to black gradient.
+ - '@b' after a map file displays the dots in black.
+ - '@r' after a map file displays the dots in red.
+ - '@g' after a map file displays the dots in green.
+ - '@m' after a map file displays the dots in magenta.
+ - Example: 'TOPPView 1.mzML + 2.mzML @bw + 3.mzML @bg'
+```
diff --git a/docs/tutorials/KNIME/KNIME-tutorial.md b/docs/tutorials/KNIME/KNIME-tutorial.md
new file mode 100644
index 00000000..179f4184
--- /dev/null
+++ b/docs/tutorials/KNIME/KNIME-tutorial.md
@@ -0,0 +1,67 @@
+KNIME Tutorial
+==============
+
+Users can now use {term}`KNIME` in place of {term}`TOPPAS`; the later will deprecated with no support in near future.
+
+The below image shows an example workflow in {term}`KNIME`.
+
+
+
+
+## Installing OpenMS in KNIME
+
+Installation of OpenMS in {term}`KNIME` is platform-independent across Windows, MacOSX, and Linux.
+
+1. Download the latest {term}`KNIME` release from the [KNIME website](https://www.knime.com/).
+2. In the full install of {term}`KNIME` skip the following installation routine since all required plugins should be
+ installed by default. For the standard (core) installation, follow the instructions here or in the extended [user-tutorial]().
+
+ 
+
+3. In KNIME click on **Help** > **Install new Software**.
+4. Install the required KNIME File Handling nodes from the official KNIME Update Site (a standard entry in the update
+ sites). Choose the update site from the **Work with:** dropdown menu.
+
+
+ **Name**: KNIME Analytics Platform ${YOUR_KNIME_VERSION} Update Site.
+
+ **Location**: http://update.knime.org/analytics-platform/${YOUR_KNIME_VERSION}
+
+5. Filter the results for **File handling** and select the {term}`KNIME` File Handling Nodes. Click **Next** and install.
+
+ 
+
+6. Now, install the actual OpenMS pluging. Next to the **Work with:** dropdown menu, click on **Add...**. In the opening
+ dialog fill in at least one of the following additional Update Sites (if not already present):
+
+ **Recommened**:
+
+ **Name**: KNIME Community Contributions (Stable)
+
+ **Location**: http://update.knime.org/community-contributions/trusted/${YOUR_KNIME_VERSION}
+
+ **Unstable**:
+
+ **Name**: KNIME Nightly Community Contributions (trunk)
+
+ **Location**: http://update.knime.org/community-contributions/trunk
+
+7. Use the search or navigate to **KNIME Community Contributions – Bioinformatics & NGS** and select **OpenMS**. Then
+ click **Next** and follow the installation instructions. A restart of KNIME might be necessary afterward. On Windows,
+ if prompted to install additional requirements like the Microsoft Visual Studio Redistributable for the conversion
+ software ProteoWizard that is packaged with our plugin.
+8. After a restart of KNIME the OpenMS nodes will be available in your Node Repository (panel on the lower left) under
+ **Community Nodes**.
+
+## Creating workflows with KNIME
+
+Download Introduction to OpenMS in KNIME [user tutorial]() containing hands-on training material covering also basic
+usage of KNIME. See the official [KNIME Getting Started Guide](https://tech.knime.org/knime) for a more in-depth view of
+the KNIME functionality besides OpenMS.
+
+If you face any issues, please [contact us](../../contact-us.md) and specifically for the usage of OpenMS in KNIME, the
+KNIME community contribution [forum](https://tech.knime.org/forum/openms).
+
+## Creating your own Generic KNIME Nodes
+
+See the more advanced instructions on our [Getting Started]() page for developers in OpenMS API reference.
diff --git a/docs/tutorials/TOPP/TOPP-tutorial.md b/docs/tutorials/TOPP/TOPP-tutorial.md
new file mode 100644
index 00000000..10e6ec9b
--- /dev/null
+++ b/docs/tutorials/TOPP/TOPP-tutorial.md
@@ -0,0 +1,80 @@
+TOPP Tutorial
+============
+
+The following tutorial is an introduction to {term}`TOPP` and {term}`TOPPView`. Let's start with understanding the intent and
+concept of {term}`TOPP` compared to OpenMS. Later, we will move to the handling of {term}`TOPPView` which is our central
+GUI. Apart from viewing data, {term}`TOPPView` can also be used to analyze it using selected {term}`TOPP tools`; how-to
+is explained in the third part of the tutorial.
+
+Finally, the tutorial lists the command line interfaces for all TOPP tools contained in the release.
+
+# Concepts
+
+Let's understand the intent of [TOPP and OpenMS](topp-and-openms-introduction.md).
+
+```{toctree}
+:maxdepth: 1
+
+topp-and-openms-introduction
+```
+
+# TOPPView Main Interface
+
+In the following section, we will learn about the main features of {term}`TOPPView` and its basic uses.
+
+```{toctree}
+:maxdepth: 1
+
+toppview-introduction
+views-in-toppview
+display-modes-and-view-options
+data-analysis-in-toppview
+data-editing-in-toppview
+hotkeys-table
+```
+
+# Calling TOPP tools from TOPPView
+
+The following part of the tutorial illustrates how to interactively analyse {term}`proteomics` data using {term}`TOPP tools` from
+within {term}`TOPPView`.
+
+```{toctree}
+:maxdepth: 1
+
+smoothing-raw-data
+subtracting-a-baseline-from-a-spectrum
+picking-peaks
+feature-detection-on-centroided-data
+```
+
+# Advanced Users: Tips & Tricks
+
+Read [TOPP for Advanced Users](topp-for-advanced-users.md) to know more about how to use advanced functionalities,
+increasing productivity.
+
+```{toctree}
+:maxdepth: 1
+
+topp-for-advanced-users
+```
+
+# Scripting with TOPP
+
+This part of the tutorial gives a detailed overview of the most important {term}`TOPP tools`. First, some basics needed
+for every {term}`TOPP tool` are explained, then several example pipelines are shown.
+
+```{toctree}
+:maxdepth: 1
+
+general-introduction
+file-handling
+profile-data-processing
+calibration
+map-alignment
+feature-detection
+feature-grouping
+consensus-peptide-identification
+peptide-property-prediction
+quality-control
+conversion-between-openms-xml-formats-and-text-formats
+```
diff --git a/docs/tutorials/TOPP/calibration.md b/docs/tutorials/TOPP/calibration.md
new file mode 100644
index 00000000..fa65e17b
--- /dev/null
+++ b/docs/tutorials/TOPP/calibration.md
@@ -0,0 +1,25 @@
+Calibration
+===========
+
+OpenMS offer two calibration methods: an internal and an external calibration. Both can handle peak data as well as
+profile data. To calibrate profile data, a peak picking step is necessary, the important parameters can be set via the
+ini-file. If you have already picked data, don't forget the `-peak_data` flag.
+
+The external calibration (**TOFCalibration**) is used to convert flight times into m/z values with the help of external
+calibrant spectra containing e.g. a polymer like polylysine. For the calibrant spectra, the calibration constants the
+machine uses need to be known as well as the expected masses. Then a quadratic function is fitted to convert the flight
+times into m/z-values.
+
+The internal calibration (**InternalCalibration**) uses reference masses in the spectra to correct the m/z-values
+using a linear function.
+
+In a typical setting one would first pick the TOF-data, then perform the TOFCalibration and then the InternalCalibration:
+
+```bash
+PeakPickerWavelet -in raw_tof.mzML -out picked_tof.mzML -ini pp.ini
+TOFCalibration -in picked_tof.mzML -out picked.mzML -ext_calibrants ext_cal.mzML
+ -ref_masses ext_cal_masses
+ -tof_const tof_conv_consts -peak_data
+InternalCalibration -in picked.mzML -out picked_calibrated.mzML
+ -ref_masses internal_calibrant_masses -peak_data
+```
diff --git a/docs/tutorials/TOPP/consensus-peptide-identification.md b/docs/tutorials/TOPP/consensus-peptide-identification.md
new file mode 100644
index 00000000..74658911
--- /dev/null
+++ b/docs/tutorials/TOPP/consensus-peptide-identification.md
@@ -0,0 +1,22 @@
+Consensus Peptide Identification
+===============================
+
+In order to compute a consensus identification for a HPLC-MS\MS experiment use several identification engines.
+
+OpenMS offers adapters for the following commercial and free peptide identification engines: Sequest, Mascot, OMSSA,
+PepNovo, XTandem and Inspect. The adapters allow setting the input parameters and data for the identification engine
+and return the result in the OpenMS idXML format.
+
+In order to improve the identification accuracy, several identification engines can be used and a consensus
+identification can be calculated from the results. The image below shows an example where Mascot and OMSSA results are
+fed to the **ConsensusID** tool (ConsensusID is currently usable for Mascot, OMSSA and XTandem).
+
+
+
+
+To combine quantitation and identification results:
+
+Protein/peptide identifications can be annotated to quantitation results (featureXML, consensusXML) by the **IDMapper**
+tool. The combined results can then be exported by the **TextExporter** tool:
+
+[Conversion between OpenMS XML formats and text formats](conversion-between-openms-xml-formats-and-text-formats.md).
diff --git a/docs/tutorials/TOPP/conversion-between-openms-xml-formats-and-text-formats.md b/docs/tutorials/TOPP/conversion-between-openms-xml-formats-and-text-formats.md
new file mode 100644
index 00000000..57d2af30
--- /dev/null
+++ b/docs/tutorials/TOPP/conversion-between-openms-xml-formats-and-text-formats.md
@@ -0,0 +1,64 @@
+Conversion Between OpenMS XML Formats and Text Formats
+=====================================================
+
+## Export of OpenMS XML formats
+
+As TOPP offers no functionality for statistical analysis, this step is normally done using external statistics packages.
+In order to export the OpenMS XML formats into an appropriate format for these packages the TOPP **TextExporter** can be
+used.
+
+It converts the the following OpenMS XML formats to text files:
+
+- featureXML
+- idXML
+- consensusXML
+
+The use of the `TextExporter` is is very simple:
+
+`TextExporter -in infile.idXML -out outfile.txt`
+
+## Import of feature data to OpenMS
+
+OpenMS offers a lot of visualization and analysis functionality for feature data.
+Feature data in text format, e.g. from other analysis tools, can be imported using the `TextImporter`. The default
+mode accepts comma separated values containing the following columns: RT, m/z, intensity. Additionally meta data
+columns may follow. If meta data is used, meta data column names have to be specified in a header line. Without headers:
+
+```bash
+1201 503.123 1435000
+1201 1006.246 1235200
+```
+
+Or with headers:
+
+```bash
+RT m/z Int isHeavy myMeta
+1201 503.123 1435000 true 2
+1201 1006.246 1235200 maybe 1
+```
+
+Example invocation:
+
+`TextImporter -in infile.txt -out outfile.featureXML`
+
+The tool also supports data from msInspect,SpecArray and Kroenik(Hardkloer sibling), just specify the `-mode` option
+accordingly.
+
+## Import of protein/peptide identification data to OpenMS
+
+Peptide/protein identification data from several identification engines can be converted to idXML format using the
+**IDFileConverter** tool.
+
+It can currently read the following formats:
+- Sequest output folder
+- pepXML file
+- idXML file
+
+It can currently write the following formats:
+
+- pepXML
+- idXML
+
+This example shows how to convert pepXML to idXML:
+
+`IDFileConverter -in infile.pepXML -out outfile.idXML`
diff --git a/docs/tutorials/TOPP/data-analysis-in-toppview.md b/docs/tutorials/TOPP/data-analysis-in-toppview.md
new file mode 100644
index 00000000..f7732c08
--- /dev/null
+++ b/docs/tutorials/TOPP/data-analysis-in-toppview.md
@@ -0,0 +1,40 @@
+Data Analysis in TOPPView
+=========================
+
+{term}`TOPPView` also offers limited data analysis capabilities for single layers, which will be illustrated in the following
+sections. The functionality presented here can be found in the **Tools** menu:
+
+
+
+## TOPP Tools
+
+Single {term}`TOPP tools` can be applied to the data of the currently selected layer or to the visible data of the current layer.
+The following example image shows the TOPP tools dialog:
+
+
+
+To apply a {term}`TOPP tool`, follow the instructions below:
+
+1. Select a {term}`TOPP tool` and if necessary a type.
+2. Specify the command line option of the tool, that takes the input file name.
+3. Specify the command line option of the tool, that takes the output file name.
+4. Set the algorithm parameters manually or load them from an INI file.
+
+## Metadata
+
+One can access the metadata, the layer is annotated with. This data comprises e.g. contact person, instrument description
+and sample description.
+
+
+
+```{tip}
+Identification data, e.g. from a {term}`Mascot` run, can be annotated to the spectra or features, too. After
+annotation, this data is listed in the metadata.
+```
+
+## Statistics
+
+Statistics about peak/feature intensities and peak meta information can be displayed. For intensities, it is possible to
+display an additional histogram view.
+
+
diff --git a/docs/tutorials/TOPP/data-editing-in-toppview.md b/docs/tutorials/TOPP/data-editing-in-toppview.md
new file mode 100644
index 00000000..85666f5e
--- /dev/null
+++ b/docs/tutorials/TOPP/data-editing-in-toppview.md
@@ -0,0 +1,11 @@
+Data Editing in TOPPView
+========================
+
+{term}`TOPPView` offers editing functionality for feature layers.
+
+After enabling the feature editing mode in the context menu of the feature layer, the following actions can be performed:
+
+- Features can be dragged with the mouse in order to change the {term}`m/z` and {term}`RT` position.
+- The position, intensity and charge of a feature can be edited by double-clicking a feature.
+- Features can be created by double-clicking the layer background.
+- Features can be removed by selecting them and pressing the DEL key.
diff --git a/docs/tutorials/TOPP/display-modes-and-view-options.md b/docs/tutorials/TOPP/display-modes-and-view-options.md
new file mode 100644
index 00000000..75b4eac6
--- /dev/null
+++ b/docs/tutorials/TOPP/display-modes-and-view-options.md
@@ -0,0 +1,52 @@
+Display Modes and View Options in TOPPView
+==========================================
+
+All of the views support several display modes and view options.
+Display modes determine how intensities are displayed. View options configure the view.
+
+
+
+## Display Modes
+
+Intensity display modes determine the way peak intensities are displayed.
+
+### Linear
+
+Normal display mode.
+
+### Percentage
+
+In this display mode the intensities of each dataset are normalized with the maximum intensity of the dataset. This is
+especially useful in order to visualize several datasets that have large intensity differences. If only one dataset is
+opened it corresponds to the normal mode.
+
+### Snap to Maximum Intensity
+
+In this mode the maximum currently displayed intensity is treated as if it was the maximum overall intensity.
+
+## View Options
+
+View options configure the view of the current layer.
+
+### 1D
+
+Switching between raw data and peak mode.
+
+### 2D (Peaks)
+
+`MS/MS` precursor peaks can be highlighted.
+Projections to **m/z** and **RT** axis can be shown.
+
+### 2D (Features)
+
+Overall convex hull of the features can be displayed.
+Convex hulls of mass traces can be displayed (if available).
+A feature identifier, consisting of the feature index and an optional label can be displayed.
+
+### 2D (Consensus)
+
+The elements of a consensus feature can be displayed.
+
+### 3D
+
+Currently there are no options for 3D view.
diff --git a/docs/tutorials/TOPP/feature-detection-on-centroided-data.md b/docs/tutorials/TOPP/feature-detection-on-centroided-data.md
new file mode 100644
index 00000000..b837bd6c
--- /dev/null
+++ b/docs/tutorials/TOPP/feature-detection-on-centroided-data.md
@@ -0,0 +1,31 @@
+Feature Detection on Centroided Data
+====================================
+
+To quantify peptide features, TOPP offers the **FeatureFinder** tools. In this section the **FeatureFinderCentroided**
+is used, which works only on centroided data. There are other FeatureFinders available that also work on profile data.
+
+For this example the file `LCMS-centroided.mzML` from the examples data is used (**File** > **Open example data**). In order
+to adapt the algorithm to the data, some parameters have to be set.
+
+## Intensity
+
+The algorithm estimates the significance of peak intensities in a local environment. Therefore, the HPLC-MS map is
+divided into `n` times `n` regions. Set the `intensity:bins` parameter to `10` for the whole map. For a small region, set
+it to `1`.
+
+## Mass trace
+
+For the mass traces, define the number of adjacent spectra in which a mass has to occur (`mass_trace:min_spectra`). In
+order to compensate for peak picking errors, missing peaks can be allowed (`mass_trace:max_missing`) and a tolerated
+mass deviation must be set (`mass_trace:mz_tolerance`).
+
+## Isotope pattern
+
+The expected isotopic intensity pattern is estimated from an averagene amino acid composition. The algorithm searches
+all charge states in a defined range (`isotopic_pattern:change_min` to `isotopic_pattern:change_max`). Just as for mass
+traces, a tolerated mass deviation between isotopic peaks has to be set (`isotopic_pattern:mz_tolerance`).
+
+The image shows the centroided peak data and the found peptide features. The used parameters can be found in the TOPP
+tools dialog.
+
+
diff --git a/docs/tutorials/TOPP/feature-detection.md b/docs/tutorials/TOPP/feature-detection.md
new file mode 100644
index 00000000..da9369bf
--- /dev/null
+++ b/docs/tutorials/TOPP/feature-detection.md
@@ -0,0 +1,71 @@
+Feature Detection
+=================
+
+For quantitation, the **FeatureFinder** tools are used. They extract the features from profile data or centroided data.
+TOPP offers different types of **FeatureFinders**:
+
+## FeatureFinderIsotopeWavelet
+
+### Description
+
+The algorithm has been designed to detect features in raw MS data sets. The current implementation is only able to
+handle MS1 data. An extension handling also tandem MS spectra is under development. The method is based on the **isotope
+wavelet**, which has been tailored to the detection of isotopic patterns following the averagine model. For more
+information about the theory behind this technique, please refer to Hussong et al.: "Efficient Analysis of Mass
+Spectrometry Data Using the Isotope Wavelet" (2007).
+
+```{attention}
+This algorithm features no "modelling stage", since the structure of the isotopic pattern is explicitly
+coded by the wavelet itself. The algorithm also works for 2D maps (in combination with the so-called *sweep-line*
+technique (Schulz-Trieglaff et al.: "A Fast and Accurate Algorithm for the Quantification of Peptides from Mass
+Spectrometry Data" (2007))). The algorithm could originally be executed on (several) high-speed CUDA graphics cards.
+Tests on real-world data sets revealed potential speedups beyond factors of 200 (using 2 NVIDIA Tesla cards in parallel).
+Support for CUDA was removed in [OpenMS]() due to maintenance overhead. Please refer to Hussong et al.: "Highly
+accelerated feature detection in proteomics data sets using modern graphics processing units" (2009) for more details on
+the implementation.
+```
+
+### Seeding
+
+Identification of regions of interest by convolving the signal with the wavelet function. A score, measuring the
+closeness of the transform to a theoretically determined output function, finally distinguishes potential features from
+noise.
+
+### Extension
+
+The extension is based on the sweep-line paradigm and is done on the fly after the wavelet transform.
+
+### Modelling
+
+None (explicitly done by the wavelet).
+
+See the `FeatureFinderAlgorithmIsotopeWavelet` class documentation for a parameter list.
+
+## FeatureFinderCentroided
+
+### Description
+
+This is an algorithm for feature detection based on peak data. In contrast to the other algorithms, it is based on
+peak/stick data, which makes it applicable even if no profile data is available. Another advantage is its speed due to
+the reduced amount of data after peak picking.
+
+### Seeding
+
+It identifies interesting regions by calculating a score for each peak based on
+
+- the significance of the intensity in the local environment.
+- RT dimension: the quality of the mass trace in a local RT window.
+- m/z dimension: the quality of fit to an averagine isotope model.
+
+### Extension
+
+The extension is based on a heuristics – the average slope of the mass trace for RT dimension, the best fit to averagine
+model in m/z dimension.
+
+### Modelling
+
+In model fitting, the retention time profile (Gaussian) of all mass traces is fitted to the data at the same time. After
+fitting, the data is truncated in RT and m/z dimension. The reported feature intensity is based on the fitted model,
+rather than on the (noisy) data.
+
+See the `FeatureFinderAlgorithmPicked` class documentation for a parameter list.
diff --git a/docs/tutorials/TOPP/feature-grouping.md b/docs/tutorials/TOPP/feature-grouping.md
new file mode 100644
index 00000000..9d8ac124
--- /dev/null
+++ b/docs/tutorials/TOPP/feature-grouping.md
@@ -0,0 +1,35 @@
+Feature Grouping
+================
+
+In order to quantify differences across maps (label-free) or within a map (isotope-labeled), groups of corresponding
+features have to be found. The **FeatureLinker** TOPP tools support both approaches. These groups are represented by
+consensus features, which contain information about the constituting features in the maps as well as average position,
+intensity, and charge.
+
+## Isotope-labeled quantitation
+
+To differentially quantify the features of an isotope-labeled HPLC-MS map, follow the listed steps:
+
+1. The first step in this pipeline is to find the features of the HPLC-MS map. The FeatureFinder applications calculate
+ the features from profile data or centroided data.
+2. In the second step, the labeled pairs (e.g. light/heavy labels of ICAT) are determined by the **FeatureLinkerLabeled**
+ application. **FeatureLinkerLabeled** first determines all possible pairs according to a given optimal shift and
+ deviations in RT and m/z. Then it resolves ambiguous pairs using a greedy-algorithm that prefers pairs with a higher
+ score. The score of a pair is the product of:
+
+ - feature quality of feature 1
+ - feature quality of feature 2
+ - quality measure for the shift (how near is it to the optimal shift)
+
+ 
+
+## Label-free quantitation
+
+To differentially quantify the features of two or more label-free HPLC-MS map.
+
+
+
+```{tip}
+This algorithm assumes that the retention time axes of all input maps are very similar. To correct for
+retention time distortions, please have a look at [Map alignment](map-alignment.md).
+```
diff --git a/docs/tutorials/TOPP/file-handling.md b/docs/tutorials/TOPP/file-handling.md
new file mode 100644
index 00000000..9394d040
--- /dev/null
+++ b/docs/tutorials/TOPP/file-handling.md
@@ -0,0 +1,58 @@
+File Handling
+============
+
+## General information about peak and feature maps
+
+For general information about a peak or feature map, use the **FileInfo** tool.
+
+- It can print RT, m/z and intensity ranges, the overall number of peaks, and the distribution of MS levels.
+- It can print a statistical summary of intensities.
+- It can print some meta information.
+- It can validate XML files against their schema.
+- It can check for corrupt data in peak files See the `FileInfo –help` for details.
+
+## Problems with input files
+
+If you are experiencing problems while processing an XML file, check if the file does validate against the XML schema:
+
+`FileInfo -v -in infile.mzML`
+
+Validation is available for several file formats including mzML, mzData, mzXML, featureXML and idXML.
+
+Another frequently-occurring problem is corrupt data. You can check for corrupt data in peak files with `FileInfo` as well:
+
+`FileInfo -c -in infile.mzML`
+
+## Converting your files to mzML
+
+The TOPP tools work only on the HUPO-PSI `mzML` format. If you need to convert *mzData*, *mzXML* or *ANDI/MS* data to
+*mzML*, use the FileConverter, e.g.
+
+`FileConverter -in infile.mzXML -out outfile.mzML`
+
+For format names as file extension, the tool derives the format from the extension. For other extensions, the file
+formats of the input and output file can be given explicitly.
+
+## Converting between DTA and mzML
+
+Sequest DTA files can be extracted from a mzML file using the `DTAExtractor`:
+
+`DTAExtractor -in infile.mzML -out outfile`
+
+The retention time of a scan, the precursor mass-to-charge ratio (for MS/MS scans) and the file extension are appended
+to the output file name.
+
+To combine several files (e.g. DTA files) to an `mzML` file use the `FileMerger`:
+
+`FileMerger -in infile_list.txt -out outfile.mzML`
+
+The retention times of the scans can be generated, taken from the `infile_list.txt` or can be extracted from the DTA
+file names. See the FileMerger documentation for details.
+
+## Extracting part of the data from a file
+
+To extract part of the data from an `mzML` file, use the `FileFilter` tool. It allows filtering for RT, `m/z` and
+intensity range or for MS level. To extract the MS/MS scans between retention time `100` and `1500`, use the following
+command:
+
+`FileFilter -in infile.mzML -levels 2 -rt 100:1500 -out outfile.mzML`
diff --git a/docs/tutorials/TOPP/general-introduction.md b/docs/tutorials/TOPP/general-introduction.md
new file mode 100644
index 00000000..12266c7d
--- /dev/null
+++ b/docs/tutorials/TOPP/general-introduction.md
@@ -0,0 +1,141 @@
+General Introduction
+====================
+
+This tutorial will gives a brief overview of the most important **TOPP** tools. First, some basics that are required for
+every TOPP tool, then there are several example pipelines explained.
+
+## File formats
+
+The TOPP tools use the HUPO-PSI standard format mzML 1.1.0 as input format. In order to convert other open formats
+(mzData, mzXML, DTA, ANDI/MS) to mzML, a file converter is provided by TOPP.
+
+Proprietary MS machine formats are not supported. To convert these formats to mzML, mzData or mzXML, please have a look
+at the [SASHIMI project page](http://tools.proteomecenter.org/wiki/index.php) or contact your MS machine vendor.
+
+mzML covers only the output of a mass spectrometry experiment. For further analysis of the data several other file
+formats are needed. The main file formats used by TOPP are:
+
+1. **mzML**: The HUPO-PSI standard format for mass spectrometry data.
+2. **featureXML**: The OpenMS format for quantitation results.
+3. **consensusXML**: The OpenMS format for grouping features in one map or across several maps.
+4. **idXML**: The OpenMS format for protein and peptide identification.
+
+Documented schemas of the OpenMS formats can be found [here](https://github.com/OpenMS/OpenMS/tree/develop/share/OpenMS/SCHEMAS).
+
+`idXML` files and `consensusXML` files created by OpenMS can be visualized in a web browser. XSLT stylesheets are used to
+transform the XML to HTML code. The stylesheets are contained in the `OpenMS/share/OpenMS/XSLT/` folder of your OpenMS
+installation.
+
+To view the file on the computer with the OpenMS installation, open it in your browser.
+
+To copy the file to another computer, copy the XSLT stylesheet to that computer and change the second line in the XML
+file. The following example shows how to change the stylesheet location for an idXML file. Change the `PATH` in the
+line as:
+
+```
+
+```
+
+to the folder where the stylesheet resides.
+
+## Common arguments of the TOPP tools
+
+The command line and INI file parameters of the TOPP tools vary due to the different tasks of the TOPP tools. However,
+all TOPP tools share this common interface:
+
+1. **-ini** ``: Use the given TOPP INI file.
+2. **-log** ``: Location of the log file (default: `TOPP.log`).
+3. **-instance** ``: Instance number in the TOPP INI file (default: `1`).
+4. **-debug** ``: Sets the debug level (default: `0`).
+5. **-write_ini** ``: Writes an example INI file.
+6. **-no_progress**: Disables progress logging to command line.
+7. **–help**: Shows a help page for the command line and INI file options.
+8. **–helphelp**: Shows a detailed (including advanced parameters) help page for the command line and INI file options.
+
+## TOPP INI files
+
+Each TOPP tool has its own set of parameters which can be specified at the command line. However, a more convenient
+(and persistent) way to handle larger sets of parameters is to use TOPP INI files. TOPP INI files are XML-based and
+can contain the configuration of one or several TOPP tools.
+
+The following examples will give an overview of how TOPP tools can be chained in order to create analysis pipelines. INI
+files are the recommended way to store all settings of such a pipeline in a single place.
+
+```{attention}
+The issue of finding suitable parameters for the tools is not addressed here. For problems during the
+execution of the example pipelines on the data, adapt the parameters. Have a look at the documentation of the
+corresponding TOPP tool in that case.
+```
+
+### Parameter documentation
+
+General documentation of a TOPP tool and documentation for the command line parameters, can be displayed using the
+command line flag `-help`. Some TOPP tools also have subsections of parameters that are internally handed to an
+algorithm. The documentation of these subsections is not displayed with `–help`. It is however displayed in
+**INIFileEditor**.
+
+
+
+In the [INIFileEditor](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_INIFileEditor.html), click on the respective parameter, the documentation of the parameters is displayed in the window at the bottom.
+
+### Updating an INI file for a TOPP tool or a whole TOPPAS pipeline
+
+For an old INI file which does not work for a newer [OpenMS]() version (due to renamed/removed or new) parameters, you
+can rescue parameters whose name did not change into the new version by using [INIUpdater](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/UTILS_INIUpdater.html) tool by calling it with (a list of) outdated
+INI and/or TOPPAS files. See the INIUpdater tool description for details. This will remove invalid parameters and add
+new parameters (if available) while retaining values for unchanged parameters. As an alternative to the INIUpdater, use
+the command line by calling the TOPP tool from which the ini originated and combining `-write_ini` and `-ini`, e.g.,
+
+```
+FileInfo -ini old_fi.ini -write_ini fi.ini
+```
+
+This will transfer all values of parameters from `old_fi.ini` which are still valid to the `fi.ini`.
+
+### General structure of an INI file
+
+An INI file is always enclosed by the `` tag. Inside this tag, a tree-like hierarchy is created with ``
+tags that represent *sections* and `` tags, each of which stores one of the parameters. The first two level of the
+hierarchy have a special meaning.
+
+**Example**: Below is the content of an INI file for **FileFilter**.
+
+Several parameter sets for a TOPP tool can be specified in a *tool* section. The tool section is always named after the
+program itself, in this case `FileFilter`.
+
+- In order to make storing several parameter sets for the same tool in one INI file possible, , the tool section
+contains one or several *numbered instance subsections* ('1', '2', ...). These numbers are the instance numbers which
+can be specified using the `-instance` command line argument. (Remember the default is `1`).
+- Within each instance section, the actual parameters of the TOPP tool are given. INI files for complex tools can
+contain nested subsections in order to group related parameters.
+- If a parameter is not found in the instance section, the *tool-specific common section* is considered.
+- Finally, we look if the *general common section* contains a value for the parameter.
+
+As an example, let's call the `FileFilter` tool with the INI file given below and instance number `2`. The `FileFilter`
+parameters `rt` and `mz` are looked up by the tool. *mz* can be found in section **FileFilter** - `2`. `rt` is not
+specified in this section, thus the `common` **FileFilter** section is checked first, where it is found in our example.
+When looking up the `debug` parameter, the tool would search the instance section and tool-specific common section
+without finding a value. Finally, the *general common section* would be checked, where the debug level is specified.
+
+```xml
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+
+```
diff --git a/docs/tutorials/TOPP/hotkeys-table.md b/docs/tutorials/TOPP/hotkeys-table.md
new file mode 100644
index 00000000..bb322e63
--- /dev/null
+++ b/docs/tutorials/TOPP/hotkeys-table.md
@@ -0,0 +1,78 @@
+TOPPView Hotkeys
+================
+
+## File handling
+
+| Hotkey | Function |
+|---------------------------------------------------------|-----------------------------------------------------|
+| CTRL + O | Open file |
+| CTRL + W | Close current window |
+| CTRL + S | Save current layer |
+| CTRL + SHIFT + S | Save visible data of current layer |
+
+
+## Navigation in the data
+
+| Hotkey | Function |
+|------------------------------------|-----------------------------------------------------|
+| CTRL | Activate zoom mode |
+| SHIFT | Activate measurement mode |
+| Arrow keys | Translate currently shown data (1D View: Shift + Left/Right moves to the next peak in sparse data)|
+|CTRL + +, CTRL + - | Move up and down in zoom history |
+| Mouse wheel | Move up and down in zoom history |
+| CTRL + G | Go to dialog |
+| Backspace | Reset zoom |
+| PageUp | Select previous layer |
+| PageDown | Select next layer |
+
+
+## Visualization options
+
+| Hotkey | Function |
+|----------------------|-----------------------------------------------------|
+| CTRL + R | Show/hide grid lines |
+| CTRL + L | Show/hide axis legends |
+| N | Intensity mode: Normal |
+| P | Intensity mode: Percentage |
+| S | Intensity mode: Snam-to-maximum |
+| I | 1D draw mode: peaks |
+| R | 1D draw mode: raw data |
+| CTRL + ALT + Home | 2D draw mode: increase minimum canvas coverage threshold (for raw peak scaling)|
+| CTRL + ALT + End | 2D draw mode: decrease minimum canvas coverage threshold (for raw peak scaling) |
+| CTRL + ALT + + | 2D draw mode: increase maximum point size (for raw peak scaling) |
+| CTRL + ALT + - | 2D draw mode: decrease maximum point size (for raw peak scaling) |
+
+```{tip}
+Home on macOS keyboards is also Fn + ArrowLeft. End on macOSX keyboards is also Fn + ArrowRight.
+```
+
+## Annotations in 1D view
+
+| Hotkey | Function |
+|------------------------------------------|-----------------------------------------------------|
+| CTRL + A | Select all annotations of the current layer |
+| DEL | Delete all currently selected annotations |
+
+
+## Advanced
+
+| Hotkey | Function |
+|-----------------------------------------------------|---------------------------------------------------------|
+| CTRL + T | Apply TOPP tool to the current layer |
+| CTRL + SHIFT + T | Apply TOPP tool to the visible data of the current layer|
+| F4 | Re-run TOPP tool |
+| CTRL + M | Show layer meta information |
+| CTRL + I | Annotate with identification results |
+| 1 | Show precursor peaks (2D peak layer) |
+| 2 | Show projections (2D peak layer) |
+| 5 | Show overall convex hull (2D feature layer) |
+| 6 | Show all convex hulls (2D feature layer) |
+| 7 | Show numbers and labels (2D feature layer) |
+| 9 | Show consensus elements (2D consensus layer) |
+
+## Help
+
+| Hotkey | Function |
+|----------------------------------------|-----------------------------------------------------|
+| F1 | Show TOPPView online tutorial |
+| SHIFT + F1 | Activate *What's this?* mode |
diff --git a/docs/tutorials/TOPP/map-alignment.md b/docs/tutorials/TOPP/map-alignment.md
new file mode 100644
index 00000000..c88dc781
--- /dev/null
+++ b/docs/tutorials/TOPP/map-alignment.md
@@ -0,0 +1,21 @@
+Map Alignment
+============
+
+## Map alignment
+
+The goal of map alignment is to transform different HPLC-MS maps (or derived maps) to a common retention time axis. It
+corrects for shifted and scaled retention times, which may result from changes of the chromatography.
+
+The different **MapAligner** tools take `n` input maps, de-warp them and store the `n` de-warped maps. The following
+image shows the general procedure:
+
+
+
+There are different map alignment tools available. The following table gives a rough overview of them:
+
+| Tool | Applicable To | Description |
+|-------------|---------------|-------------|
+| `MapAlignerPoseClustering` | feature maps, peak maps | This algorithm does a star-wise alignment of the input data. The center of the star is the map with most data points. All other maps are then aligned to the center map by estimating a linear transformation (shift and scaling) of retention times. The transformation is estimated using a pose clustering approach as described in doi:10.1093/bioinformatics/btm209 |
+| `MapAlignerIdentification` | feature maps, consensus maps, identifications | This algorithm utilizes peptide identifications, and is thus applicable to files containing peptide IDs (idXML, annotated featureXML/consensusXML). It finds peptide sequences that different input files have in common and uses them as points of correspondence. From the retention times of these peptides, transformations are computed that convert each file to a consensus time scale. |
+| `MapAlignerSpectrum` | peak maps | This *experimental* algorithm uses a dynamic-programming approach based on spectrum similarity for the alignment. The resulting retention time mapping of dynamic-programming is then smoothed by fitting a spline to the retention time pairs. |
+| `MapRTTransformer` | peak maps, feature maps, consensus maps, identifications | This algorithm merely *applies* a set of transformations that are read from files (in TransformationXML format). These transformations might have been generated by a previous invocation of a MapAligner tool. For example, compute a transformation based on identifications and then apply it to the features or raw data. The transformation file format is not very complicated, so it is relatively easy to write (or generate) the transformation files |
diff --git a/docs/tutorials/TOPP/peptide-property-prediction.md b/docs/tutorials/TOPP/peptide-property-prediction.md
new file mode 100644
index 00000000..c46ec173
--- /dev/null
+++ b/docs/tutorials/TOPP/peptide-property-prediction.md
@@ -0,0 +1,65 @@
+Peptide Property Prediction
+===========================
+
+Train a model for retention time prediction as well as for the prediction of proteotypic peptides.
+
+Two applications has been described in the following publications: Nico Pfeifer, Andreas Leinenbach, Christian G. Huber
+and Oliver Kohlbacher Statistical learning of peptide retention behavior in chromatographic separations: A new
+kernel-based approach for computational proteomics. BMC Bioinformatics 2007, 8:468 Nico Pfeifer, Andreas Leinenbach,
+Christian G. Huber and Oliver Kohlbacher Improving Peptide Identification in Proteome Analysis by a Two-Dimensional
+Retention Time Filtering Approach J. Proteome Res. 2009, 8(8):4109-15.
+
+The predicted retention time can be used in `IDFilter` to filter out false identifications. For data from several
+identification runs:
+
+1. Align the data using MapAligner.
+2. Use the various identification wrappers like `MascotAdapter`, `OMSSAAdapter`, ... to get the identifications.
+3. To train a model using `RTModel` use `IDFilter` for one of the runs to get the high scoring identifications (40 to 200
+ distinct peptides should be enough).
+4. Use `RTModel` as described in the documentation to train a model for these spectra. With this model, use `RTPredict`
+ to predict the retention times for the remaining runs. The predicted retention times are stored in the idXML files.
+ These predicted retention times can then be used to filter out false identifications using the `IDFilter` tool.
+
+A typical sequence of TOPP tools would look like this:
+
+```bash
+MapAligner -in Run1.mzML,...,Run4.mzML -out Run1_aligned.mzML,...,Run4_aligned.mzML
+MascotAdapter -in Run1_aligned.mzML -out Run1_aligned.idXML -ini Mascot.ini
+MascotAdapter -in Run2_aligned.mzML -out Run2_aligned.idXML -ini Mascot.ini
+MascotAdapter -in Run3_aligned.mzML -out Run3_aligned.idXML -ini Mascot.ini
+MascotAdapter -in Run4_aligned.mzML -out Run4_aligned.idXML -ini Mascot.ini
+IDFilter -in Run1_aligned.idXML -out Run1_best_hits.idXML -pep_fraction 1 -best_hits
+RTModel -in Run1_best_hits.idXML -out Run1.model -ini RT.ini
+RTPredict -in Run2_aligned.idXML -out Run2_predicted.idXML -svm_model Run1.model
+RTPredict -in Run3_aligned.idXML -out Run3_predicted.idXML -svm_model Run1.model
+RTPredict -in Run4_aligned.idXML -out Run4_predicted.idXML -svm_model Run1.model
+IDFilter -in Run2_predicted.mzML -out Run2_filtered.mzML -rt_filtering
+IDFilter -in Run3_predicted.mzML -out Run3_filtered.mzML -rt_filtering
+IDFilter -in Run4_predicted.mzML -out Run4_filtered.mzML -rt_filtering
+```
+
+For a file with certainly identified peptides used to train a model for RT prediction, use the IDs. Therefore, the file
+has to have one peptide sequence together with the RT per line (separated by one tab or space). This can then be loaded
+by `RTModel` using the `-textfile_input` flag:
+
+```bash
+RTModel -in IDs_with_RTs.txt -out IDs_with_RTs.model -ini RT.ini -textfile_input
+```
+
+The likelihood of a peptide to be proteotypic can be predicted using `PTModel` and `PTPredict`.
+
+Assume we have a file `PT.idXML` which contains all proteotypic peptides of a set of proteins. Lets also assume, we have
+a fasta file containing the amino acid sequences of these proteins called `mixture.fasta`. To be able to train `PTPredict`,
+negative peptides (peptides, which are not proteotypic) are required. Therefore, one can use the Digestor, which is
+located in the `APPLICATIONS/UTILS/` folder together with the `IDFilter`:
+
+```bash
+Digestor -in mixture.fasta -out all.idXML
+IDFilter -in all.idXML -out NonPT.idXML -exclusion_peptides_file PT.idXML
+
+```
+
+In this example the proteins are digested in silico and the non proteotypic peptides set is created by subtracting all
+proteotypic peptides from the set of all possible peptides. Then, train `PTModel`:
+
+`PTModel -in_positive PT.idXML -in_negative NonPT.idXML -out PT.model -ini PT.ini`
diff --git a/docs/tutorials/TOPP/picking-peaks.md b/docs/tutorials/TOPP/picking-peaks.md
new file mode 100644
index 00000000..8a00b905
--- /dev/null
+++ b/docs/tutorials/TOPP/picking-peaks.md
@@ -0,0 +1,26 @@
+Picking Peaks
+============
+
+For low resolution data, consider to smooth the data first ([Smoothing raw data](smoothing-raw-data.md)) and subtract
+the baseline ([Subtracting a baseline from a spectrum](subtracting-a-baseline-from-a-spectrum.md)) before peak picking.
+
+There are two types of PeakPickers: the **PeakPickerWavelet** and one especially suited for high resolution data
+(**PeakPickerHiRes**). This tutorial explains the PeakPickerWavelet. Use the file `peakpicker_tutorial_2.mzML` from the
+examples data (select **File** > **Open example data**).
+
+The main parameters are the peak width and the minimal signal to noise ratio for a peak to be picked. If you don't know
+the approximate `fwhm` of peaks, use the estimation included in the PeakPickerWavelet, set the flag `estimate_peak_width`
+to `true`. After applying the PeakPickerWavelet, observe which peak width was estimated and used for peak picking in the
+log window.
+
+To estimate the peak width, use the measuring tool ([Action Modes and Their Uses](views-in-toppview.md#action-modes-and-their-uses)) to determine
+the fwhm of one or several representative peaks.
+
+If the peak picker delivers only a few peaks even though the `peak_with` and `signal_to_noise` parameters are set to
+good values, consider changing the advanced parameter `fwhm_lower_bound_factor` to a lower value. All peaks with a lower
+`fwhm` than `fwhm_lower_bound_factor` \* `peak_width` are discarded.
+
+The following image shows a part of the spectrum with the picked peaks shown in green, the estimated peak width in the
+log window and the measured peak width.
+
+
diff --git a/docs/tutorials/TOPP/profile-data-processing.md b/docs/tutorials/TOPP/profile-data-processing.md
new file mode 100644
index 00000000..7c45c122
--- /dev/null
+++ b/docs/tutorials/TOPP/profile-data-processing.md
@@ -0,0 +1,63 @@
+Profile data processing
+======================
+
+## Introduction
+
+To find all peaks in the profile data:
+
+1. Eliminate noise using a **NoiseFilter**.
+2. The now smoothed profile data can be further processed by subtracting the baseline with the **BaselineFilter**.
+3. Then use one of the **PeakPickers** to find all peaks in the baseline-reduced profile data.
+
+
+
+There are two different smoothing filters: NoiseFilterGaussian and NoiseFilterSGolay. To use the Savitzky Golay filter,
+or the **BaselineFilter** with non equally spaced profile data, e.g. TOF data, you have to generate equally spaced data
+using the **Resampler** tool.
+
+## Picking peaks with a PeakPicker
+
+The **PeakPicker** tools allow for picking peaks in profile data. Currently, there are two different TOPP tools
+available, PeakPickerWavelet and PeakPickerHiRes.
+
+### PeakPickerWavelet
+
+This peak picking algorithm uses the continuous wavelet transform of a raw data signal to detect mass peaks. Afterwards
+a given asymmetric peak function is fitted to the raw data and important peak parameters (e.g. `fwhm`) are extracted. In
+an optional step these parameters can be optimized using a non-linear optimization method.
+
+The algorithm is described in detail in Lange et al. (2006) Proc. PSB-06.
+
+- **Input Data**: profile data (low/medium resolution)
+- **Application**: This algorithm was designed for low and medium resolution data. It can also be applied to
+ high-resolution data, but can be slow on large datasets.
+
+See the `PeakPickerCWT` class documentation for a parameter list.
+
+### PeakPickerHiRes
+
+This peak-picking algorithm detects ion signals in raw data and reconstructs the corresponding peak shape by cubic spline
+interpolation. Signal detection depends on the signal-to-noise ratio which is adjustable by the user (see parameter
+`signal_to_noise`). A picked peak's m/z and intensity value is given by the maximum of the underlying peak spline.
+
+Please notice that this method is still **experimental** since it has not been tested thoroughly yet.
+
+- **Input Data**: profile data (high resolution)
+- **Application**: The algorithm is best suited for high-resolution MS data (FT-ICR-MS, Orbitrap). In high-resolution
+ data, the signals of ions with similar mass-to-charge ratios (m/z) exhibit little or no overlapping and therefore
+ allow for a clear separation. Furthermore, ion signals tend to show well-defined peak shapes with narrow peak width.
+ These properties facilitate a fast computation of picked peaks so that even large data sets can be processed very
+ quickly.
+
+ See the `PeakPickerHiRes` class documentation for a parameter list.
+
+## Finding the right parameters for the NoiseFilters, the BaselineFilter and the PeakPickers
+
+Finding the right parameters is not trivial. The default parameters will not work on most datasets. In order to find
+good parameters, following this procedure:
+
+1. Load the data in TOPPView.
+2. Extract a single scan from the middle of the HPLC gradient (Right click on **scan**).
+3. Experiment with the parameters until you have found the proper settings
+
+Find the **NoiseFilters**, the **BaselineFilter**, and the **PeakPickers** in **TOPPView** in the menu **Layer** > **Apply TOPP tool**.
diff --git a/docs/tutorials/TOPP/quality-control.md b/docs/tutorials/TOPP/quality-control.md
new file mode 100644
index 00000000..6f7060fd
--- /dev/null
+++ b/docs/tutorials/TOPP/quality-control.md
@@ -0,0 +1,212 @@
+Quality Control
+===============
+
+To check the quality of the data (supports label-free workflows and [IsobaricAnalyzer](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_IsobaricAnalyzer.html) output):
+
+The [QualityControl](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_QualityControl.html) TOPP tool computes and collects data which allows to compute QC metrics to check the quality of
+LC-MS data. Depending on the given input data, this tool collects data for metrics (see section [Metrics](quality-control.md#Metrics)). New
+metavalues will be added to existing data and the information will be written out in mzTab format. This mzTab file can
+then be processed using custom scripts or via the R package (see [PTXQC](https://github.com/cbielow/PTXQC/).
+
+## Workflow
+
+
+
+Find an example workflow in `OpenMS/share/OpenMS/examples/TOPPAS/QualityControl.toppas`.
+
+For data from [IsobaricAnalyzer](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_IsobaricAnalyzer.html), just provide the consensusXML as input to
+[QualityControl](https://abibuilder.informatik.uni-tuebingen.de/archive/openms/Documentation/nightly/html/TOPP_QualityControl.html). No FeatureXMLs or TrafoXMLs are required. The mzML raw file can be added as input though.
+
+## Metrics
+
+Further headings shows what each of the included metrics does, what they need to be executed and what they add to the
+data or what they return.
+
+### Input Data
+
+- **PostFDR FeatureXML**: A FeatureXML after FDR filtering.
+- **Contaminants Fasta file**: A Fasta file containing contaminant proteins.
+- **Raw mzML file**: An unchanged mzML file.
+- **InternalCalibration MzML file**: An MzML file after internal calibration.
+- **TrafoXML file**: The RT alignment function as obtained from a MapAligner.
+
+### Contaminants
+
+The Contaminants metric takes the contaminants database and digests the protein sequences with the digestion enzyme that
+is given in the featureXML. Afterwards it checks whether each of all peptide sequences of the featureXML (including the
+unassigned PeptideIdentifications) is registered in the contaminants database.
+
+#### Required input data
+
+Contaminants Fasta file, PostFDR FeatureXML
+
+#### Output
+
+**Changes in files**:
+
+- Metavalue: `is_contaminant` set to `1` or to `0` if the peptide is found in the contaminant database or not and sets
+ a `0` if not.
+
+**Other outputs**:
+
+- Returns:
+ - Contaminant ratio of all peptides.
+ - Contaminant ratio of all assigned peptides.
+ - Contaminant ratio of all unassigned peptides.
+ - Intensity ratio of all contaminants in the assigned peptides.
+ - Number of empty features, Number of all found features.
+
+### FragmentMassError
+
+The **FragmentMassError** metric computes a list of fragment mass errors for each annotated MS2 spectrum in ppm and Da.
+Afterwards it calculates the mass delta between observed and theoretical peaks.
+
+#### Required input data
+
+PostFDR FeatureXML, raw mzML file
+
+#### Output
+
+**Changes in files**:
+
+- Metavalue:
+ - `fragment_mass_error_ppm` set to the fragment mass error in parts per million.
+ - `fragment_mass_error_da` set to the fragment mass error in Dalton
+
+**Other Output**:
+
+- Returns:
+ - Average and variance of fragment mass errors in ppm
+
+### MissedCleavages
+
+The **MissedCleavages** metric counts the number of MissedCleavages per PeptideIdentification given a FeatureMap and returns
+an agglomeration statistic (observed counts). Additionally the first PeptideHit of each PeptideIdentification in the
+FeatureMap is augmented with metavalues.
+
+#### Required input data
+
+PostFDR FeatureXML
+
+#### Output
+
+**Changes in files**:
+
+- Metavalue:
+ - `missed_cleavages`
+
+**Other Output**:
+
+- Returns:
+ - Frequency map of missed cleavages as key/value pairs.
+
+### MS2IdentificationRate
+
+The **MS2IdentificationRate** metric calculates the Rate of the MS2 identification as follows: The number of all
+PeptideIdentifications are counted and that number is divided by the total number of MS2 spectra.
+
+#### Required input data
+
+PostFDR FeatureXML, raw mzML file.
+
+#### Output
+
+**Changes in files**: This metric does not change anything in the data.
+
+**Other Output**:
+
+- Returns:
+ - Number of PeptideIdentifications
+ - Number of MS2 spectra
+ - Ratio of #pepID/#MS2
+
+### MzCalibration
+
+The **MzCalibration** metric adds new metavalues to the first (best) hit of each PeptideIdentification. For this metric
+it is also possible to use this without an MzML File, but then only uncalibrated m/z error (ppm) will be reported.
+However, for full functionality a PeakMap/MSExperiment with original m/z-values before m/z calibration generated by
+InternalCalibration has to be given.
+
+#### Required input data
+
+PostFDR FeatureXML
+
+#### Output
+
+**Changes in files**:
+
+- Metavalues:
+ - `mz_raw` set to m/z value of original experiment.
+ - `mz_ref` set to m/z value of calculated reference.
+ - `uncalibrated_mz_error_ppm` set to uncalibrated m/z error in parts per million.
+ - `calibrated_mz_error_ppm` set to calibrated m/z error in parts per million.
+
+**Other Output**: No additional output.
+
+### RTAlignment
+
+The **RTAlignment** metric checks what the retention time was before the alignment and how it is after the alignment.
+These two values are added to the metavalues in the PeptideIdentification.
+
+#### Required input data
+
+PostFDR FeatureXML, trafoXML file
+
+#### Output
+
+**Changes in files**:
+
+- Metavalues:
+ - `rt_align` set to retention time after alignment.
+ - `rt_raw` set to retention time before alignment.
+
+**Other Output**: No additional output.
+
+### TIC
+
+The **TIC** metric calculates the total ion count of an MSExperiment if a bin size in RT seconds greater than 0 is given.
+All MS1 abundances within a bin are summed up.
+
+#### Required input data
+
+raw mzML file
+
+#### Output
+
+**Changes in files**: This metric does not change anything in the data.
+
+**Other Output**:
+
+- Returns:
+ - TIC chromatograms
+
+### TopNoverRT
+
+The **TopNoverRT** metric calculates the ScanEventNumber (number of the MS2 scans after the MS1 scan) and adds them as the
+new metavalue `ScanEventNumber` to the PeptideIdentifications. It finds all unidentified MS2-Spectra and adds corresponding
+`empty` PeptideIdentifications without sequence as placeholders to the unassigned PeptideIdentification list.
+Furthermore, it adds the metavalue `identified` to the PeptideIdentification.
+
+#### Required input data
+
+PostFDR FeatureXML, raw mzML file
+
+#### Output
+
+**Changes in files**:
+
+- Metavalues:
+ - `ScanEventNumber` set to the calculated value
+ - `identified` set to `+` or `-`
+
+- If provided:
+ - `FWHM` set to RT peak width for all assigned PIs
+ - `ion_injection_time` set to injection time from MS2 spectrum
+ - `activation_method` set to activation method from MS2 spectrum
+ - `total_ion_count` set to summed intensity from MS2 spectrum
+ - `base_peak_intensity` set to highest intensity from MS2 spectrum
+
+- Additionally:
+ - Adds empty PeptideIdentifications
+
+**Other Output**: No additional output.
diff --git a/docs/tutorials/TOPP/smoothing-raw-data.md b/docs/tutorials/TOPP/smoothing-raw-data.md
new file mode 100644
index 00000000..32876a08
--- /dev/null
+++ b/docs/tutorials/TOPP/smoothing-raw-data.md
@@ -0,0 +1,12 @@
+Smoothing Raw Data
+==================
+
+To smooth raw data, call one of the available NoiseFilters via the Tools-menu, (select **Tools** > **Apply TOPP tool**), then select **NoiseFilterSGolay** or **NoiseFilterGaussian** as TOPP tool (green rectangle). The parameters for the filter type can be adapted (blue rectangle). For the `Savitzky-Golay` filter, set the **frame_length** and the **polynomial_order** fitted.
+For the Gaussian filter, the gaussian width and the ppm tolerance for a flexible gaussian width depending on the `m/z`
+value can be adapted. Press **Ok** to run the selected `NoiseFilter`.
+
+
+
+The following image shows a part of the spectrum after smoothing as red line with the un-smoothed data in green.
+
+
diff --git a/docs/tutorials/TOPP/subtracting-a-baseline-from-a-spectrum.md b/docs/tutorials/TOPP/subtracting-a-baseline-from-a-spectrum.md
new file mode 100644
index 00000000..f0f9e7c1
--- /dev/null
+++ b/docs/tutorials/TOPP/subtracting-a-baseline-from-a-spectrum.md
@@ -0,0 +1,17 @@
+Subtracting a Baseline from a Spectrum
+=====================================
+
+First, load the spectrum to be analyzed in TOPPView. To use the described tools, open the tutorial data via the
+File-menu (**File** > **Open example file**, then select `peakpicker_tutorial_1.mzML`). The BaselineFilter can be called via
+the Tools-menu (**Tools** > **Apply TOPP tool**), then select **BaselineFilter** as TOPPtool (red rectangle). You can choose,
+between different types of filters (green rectangle), the one mainly used is TopHat. The other important parameter is
+the length of the structuring element (blue rectangle). The default value is `3` Thomson. Press **Ok** to start the baseline
+subtraction.
+
+
+
+The following image shows:
+- A part of the spectrum after baseline filtering as a green line.
+- The original raw data as a blue line.
+
+
diff --git a/docs/tutorials/TOPP/topp-and-openms-introduction.md b/docs/tutorials/TOPP/topp-and-openms-introduction.md
new file mode 100644
index 00000000..bbd018e2
--- /dev/null
+++ b/docs/tutorials/TOPP/topp-and-openms-introduction.md
@@ -0,0 +1,16 @@
+TOPP and OpenMS
+==============
+
+{term}`TOPP`, the OpenMS Proteomics Pipeline provides a set of computational tools that can be easily combined into analysis
+pipelines even by non-experts and then be used in proteomics workflows. These applications range from useful utilities
+file format conversion, peak picking over wrapper applications for known applications (e.g.{term}`Mascot`) to completely new
+algorithmic techniques for data reduction and data analysis. {term}`TOPP` is based on the OpenMS library and as more
+functionality is added to new OpenMS releases, {term}`TOPP` will naturally contain new or updated tools.
+
+In addition to offering a toolbox, {term}`TOPP` contains {term}`TOPPView` - the central graphical user interface - which
+allows the user to view, inspect and manipulate {term}`proteomics` data. {term}`TOPPView` reads standard formats and
+allows the user not only to view the data, but also to interactively call {term}`TOPP tools` and display the results.
+As such it is a powerful tool for interactive data manipulation.
+
+In this tutorial, let's understand in detail the capabilities of {term}`TOPPView` and the {term}`TOPP tools` using real
+data sets and standard analysis tasks.
diff --git a/docs/tutorials/TOPP/topp-for-advanced-users.md b/docs/tutorials/TOPP/topp-for-advanced-users.md
new file mode 100644
index 00000000..770ec5f6
--- /dev/null
+++ b/docs/tutorials/TOPP/topp-for-advanced-users.md
@@ -0,0 +1,35 @@
+TOPP for Advanced Users
+======================
+
+This tutorials has some advanced concepts of TOPP, which will increase productivity and easy usage.
+
+## Global database for search engine adapters
+
+In your `$HOME` directory find an `OpenMS.ini` in the `.OpenMS` subfolder. This INI file contains global parameters
+which are used by many/all TOPP tools, depending on what the parameters refer to. The `id_db_dir` parameter allows to
+specify one or more directories where FASTA files (or related, e.g., `.psq` files) are placed. Specified filename
+(without path) in an ID engine adapter, will be looked up in the current working directory. If its not found, the
+directories specified in `id_db_dir` will be searched. This allows to build scripts and/or TOPPAS pipelines which are
+portable across several computers, just adapt the OpenMS.ini once on each machine.
+
+```{tip}
+When using TOPPAS: Use an "input file" node to specify the FASTA file for several engines simultaneously.
+However, when selecting the file, TOPPAS will use the absolute pathname and the dialog will not allow to name a
+non-existing file. After selecting the file you can however edit the filename and remove the path (this will issue a
+warning which can be ignored).
+```
+
+```{note}
+This approach does not work for OpenMS MascotAdapters, as each Mascot instance has its own database
+managed internally. Make sure that the database is present on all mascot servers you're going to use, thus making the
+INI settings portable.
+```
+
+## Using external tools in workflows
+
+OpenMS supports the wrapping of external tools (like msconvert from {term}`ProteoWizard`), thus allowing to build scripts and/or
+TOPPAS pipelines containing external tools.
+
+```{seealso}
+`share/OpenMS/TOOLS/EXTERNAL/ReadMe.txt` in your local installation for details.
+```
diff --git a/docs/tutorials/TOPP/toppview-introduction.md b/docs/tutorials/TOPP/toppview-introduction.md
new file mode 100644
index 00000000..9a266acf
--- /dev/null
+++ b/docs/tutorials/TOPP/toppview-introduction.md
@@ -0,0 +1,85 @@
+TOPPView Introduction
+=====================
+
+{term}`TOPPView` is a viewer for {term}`MS` and {term}`HPLC-MS` data. It can be used to inspect files in {term}`mzML`,
+{term}`mzData`, {term}`mzXML` and several other text-based file formats.
+
+In each view, several datasets can be displayed using the layer concept. This allows visual comparison of several
+datasets as well as displaying input data and output data of an algorithm together.
+
+{term}`TOPPView` is intended for visual inspection of the data by experimentalists as well as for analysis software by
+developers.
+
+
+
+The above example image shows a 2D view of TOPPView (blue rectangle) and the corresponding [Display modes and view options](display-modes-and-view-options.md) (red rectangle). The right dock area, shows the [Layers](#layers) (yellow rectangle), the [Spectrum browser](#spectrum-browser) (magenta rectangle) and the [Data filtering](data-filtering) (green rectangle). Switch between open windows can
+be done using the tab bar (black rectangle).
+
+## Layers
+
+Each view of TOPPView supports several datasets, called layers. In the *layer manager* (dock window in the upper right
+corner), layers can be hidden and shown using the check box in front of each layer name.
+
+By clicking on a layer, this layer is selected, which is indicated by a blue background. The selected layer can be
+manipulated using the *Tools* menu.
+
+Layers can be copied by dragging them to the tab bar. If the layer is dropped on a tab, it is added to the corresponding
+window. If the layer is dropped on the tab bar but not on a tab, a new window with that layer is added.
+
+## Spectrum browser
+
+The spectra contained in a peak map can be browsed in the *spectrum browser*. It displays a tree view of all spectra in
+the current layer, where a spectrum with MS level *n* is a child of the spectrum with MS level *n - 1* that contains its
+corresponding precursor peak. For each spectrum in the list, the retention time and (for spectra with MS level > 1) the
+m/z value of the precursor peak are also shown.
+
+If a 2D or 3D window is active, double-clicking a spectrum will open it in a new 1D window. If the active window is a 1D
+view, the spectra can be browsed and the currently selected spectrum will be shown.
+
+## Data filtering
+
+TOPPView allows filtering of the displayed peak data and feature data. Peak data can be filtered according to intensity
+and meta data. Meta data is arbitrary data the peak is annotated with. Feature data can be filtered according to
+intensity, charge, quality and meta data.
+
+Data filters are managed by a dock window. Filters can be added, removed and edited through the context menu (right
+button mouse click) of the data filters window. For convenience, filters can also be edited by double-clicking them.
+
+## Command line options
+
+Several files can be opened in one layer from the command line by putting a '+' character between the file names. The
+following command opens three files in three layers of the same window:
+
+```bash
+TOPPView file1.mzML + file2.mzML + file3.mzML
+```
+
+Without the '+' the files would be opened in three different tabs.
+
+The color gradient used to display a file can be changed by adding one of several '@' commands. The following command
+opens the file with a white-to-black gradient:
+
+```bash
+TOPPView file1.mzML @bw
+```
+
+Automatic annotation of a layer with ID data (.osw files etc) can be performed by using the '!' character. This will
+treat the next filename as annotation to the previous layer. The following command opens two layers in the same tab,
+with the first layer being annotated:
+
+```bash
+TOPPView file1.mzML ! file1.idXML + file2.mzML
+```
+
+For more information on command line parameters call:
+
+```bash
+TOPPView --help
+```
+
+## Looking for help?
+
+You can display a short help text for each button and dock window of TOPPView by clicking on it in *What's this?* mode.
+*What's this?* mode can be entered using the *Help* menu.
+
+
diff --git a/docs/tutorials/TOPP/views-in-toppview.md b/docs/tutorials/TOPP/views-in-toppview.md
new file mode 100644
index 00000000..4153b0c4
--- /dev/null
+++ b/docs/tutorials/TOPP/views-in-toppview.md
@@ -0,0 +1,102 @@
+Views in TOPPView
+=================
+
+{term}`TOPPView` offers three types of views – a 1D view for {term}`spectra`, a 2D view for peak maps and feature maps,
+and a 3D view for peak maps. All three views can be freely configured to suit the individual needs of the user.
+
+## Action Modes and Their Uses
+
+All three views share a similar interface. Three action modes are supported – one for translation, one for zooming
+and one for measuring:
+
+- Translate mode
+ - It is activated by default
+ - Move the mouse while holding the mouse button down to translate the current view
+ - Arrow keys can be used to translate the view without entering translate mode (in 1D-View you can additionally
+ use Shift to jump to the next peak)
+- Zoom mode
+ - All previous zoom levels are stored in a zoom history. The zoom history can be traversed using CTRL + +/CTRL + - or the
+ mouse wheel (scroll up and down)
+ - Zooming into the data:
+ - Mark an area in the current view with your mouse, while holding the left mouse button plus the CTRL key to zoom
+ to this area.
+ - You can also use your mouse wheel to traverse the zoom history.
+ - If you have reached the end of the history, keep on pressing CTRL + + or scroll up, the current area will be
+ enlarged by a factor of `1.25`.
+ - Pressing Backspace resets the zoom and zoom history.
+- Measure mode
+ - It is activated using SHIFT.
+ - Press the left mouse button down while a peak is selected and drag the mouse to another peak to measure the
+ distance between peaks.
+ - This mode is implemented in the 1D and 2D mode.
+
+## 1D View
+
+The 1D view is used to display raw spectra or peak spectra. Raw data is displayed using a continuous line. Peak data is
+displayed using one stick per peak. The color used for drawing the lines can be set for each layer individually. The 1D
+view offers a mirror mode, where the window is vertically divided in halves and individual layers can be displayed
+either above or below the "mirror" axis in order to facilitate quick visual comparison of spectra. When a mirror view is
+active, it is possible to perform a spectrum alignment of a spectrum in the upper and one in the lower half,
+respectively. Moreover, spectra can be annotated manually. Currently, distance annotations between peaks, peak
+annotations and simple text labels are provided.
+
+The following example image shows a 1D view in mirror mode. A theoretical spectrum (lower half) has been generated using
+the theoretical spectrum generator (**Tools** > **Generate theoretical spectrum**). The mirror mode has been activated by
+right-clicking the layer containing the theoretical spectrum and selecting **Flip downward** from the layer context menu.
+A spectrum alignment between the two spectra has been performed (**Tools** > **Align spectra**). It is visualized by the red
+lines connecting aligned peaks and can be reset through the context menu. Moreover, in the example, several distances
+between abundant peaks have been measured and subsequently replaced by their corresponding amino acid residue code.
+This is done by right-clicking a distance annotation and selecting **Edit** from the context menu. Additionally, peak
+annotations and text labels have been added by right-clicking peaks and selecting **Add peak** annotation or by right
+clicking anywhere and selecting **Add Label**, respectively. Multiple annotations can be selected by holding down the
+CTRL key while clicking them. They can be moved around by dragging the mouse and deleted by pressing DEL.
+
+
+
+Through the **context menu**: of the 1D view you can:
+
+1. View/edit meta data.
+2. Save the current layer data.
+3. Change display settings.
+4. Add peak annotations or arbitrary text labels.
+5. Reset a performed alignment.
+
+## 2D View
+
+The 2D view is used to display peak maps and feature maps in a top-down view with color-coded intensities. Peaks and
+feature centroids are displayed as dots. For features, also the overall convex hull and the convex hulls of individual
+mass traces can be displayed. The color gradient used to encode for peak and feature intensities can be set for each
+layer individually.
+
+The following example image shows a small section of a peak map and the detected features in a second layer.
+
+
+
+In addition to the normal top-down view, the 2D view can display the projections of the data to the `m/z` and `RT` axis.
+This feature is mainly used to assess the quality of a feature without opening the data region in 3D view.
+
+Through the **context menu:** of the 2D view you can:
+
+1. View/edit meta data
+2. View survey/fragment scans in 1D view
+3. View survey/fragment scans meta data
+4. View the currently selected area in 3D view
+5. Save the current layer data
+6. Change display settings
+
+## 3D View
+
+The 3D view can only display peak maps. Its primary use is the closer inspection of a small region of the map, e.g. a
+single feature. In the 3D view slight intensity differences are easier to recognize than in the 2D view. The color
+gradient used to encode peak intensities, the width of the lines and the coloring mode of the peaks can be set for each
+layer individually.
+
+The following example image shows a small region of a peak map:
+
+
+
+Through the **context menu**: of the 3D view you can:
+
+1. View/edit meta data.
+2. Save the current layer data.
+3. Change display setting.
diff --git a/docs/tutorials/TOPPAS/TOPPAS-tutorial.md b/docs/tutorials/TOPPAS/TOPPAS-tutorial.md
new file mode 100644
index 00000000..123a699b
--- /dev/null
+++ b/docs/tutorials/TOPPAS/TOPPAS-tutorial.md
@@ -0,0 +1,19 @@
+TOPPAS Tutorial
+==============
+
+**TOPPAS** is a GUI tool that is used to create, edit, open, save, and run TOPP workflows. Pipelines can be created
+conveniently in a GUI. The parameters of all involved tools can be edited within TOPPAS and are also saved as part of
+the pipeline definition in the `.toppas` file.
+
+```{important}
+Active development of TOPPAS is stopped. In near future, TOPPAS will be completely deprecated. Please migrate to using
+OpenMS KNIME integration.
+```
+
+```{toctree}
+:maxdepth: 1
+
+general-introduction
+user-interface
+examples
+```
diff --git a/docs/tutorials/TOPPAS/examples.md b/docs/tutorials/TOPPAS/examples.md
new file mode 100644
index 00000000..ecec532d
--- /dev/null
+++ b/docs/tutorials/TOPPAS/examples.md
@@ -0,0 +1,83 @@
+Examples
+========
+
+The following sections explain the example pipelines TOPPAS comes with. Open them by selecting **File** > **Open example file**.
+All input files and parameters are already specified, so you can just hit **Pipeline** > **Run** (or press F5) and see what happens.
+
+## Profile data processing
+
+The file `peakpicker_tutorial.toppas` contains a simple pipeline representing a common use case: starting with profile
+data, the noise is eliminated and the baseline is subtracted. Then, PeakPickerWavelet is used to find all peaks in the
+noise-filtered and baseline-reduced profile data. This workflow is also described in the section
+[Profile data processing](../TOPP/profile-data-processing.md). The individual steps are explained in more detail in the
+TOPPView tutorial: [Smoothing raw data](../TOPP/smoothing-raw-data.md), [Subtracting a baseline from a spectrum](../TOPP/subtracting-a-baseline-from-a-spectrum.md), and [Picking peaks](../TOPP/picking-peaks.md).
+
+
+
+## Identification of E. coli peptides
+
+This section describes an example identification pipeline contained in the example directory, `Ecoli_Identification.toppas`.
+The pipeline is shipped together with a reduced example mzML file containing 139 MS2 spectra from an E. coli run on an
+Orbitrap instrument as well as an E. coli target-decoy database.
+
+Use the search engine OMSSA (Geer et al., 2004) for peptide identification. Therefore, OMSSA must be installed and the
+path to the OMSSA executable (omssacl) must be set in the parameters of the OMSSAAdapter node.
+
+- Node #1 accepts mzML files containing MS2 spectra.
+- Node #2 provides the database and is set to **recycling mode** to allow the database to be reused when there is more
+ than one input file in node #1.
+- OMSSAAdapter calls OMSSA which performs the actual search.
+- PeptideIndexer annotates for each search result whether it is a target or a decoy hit.
+- FalseDiscoveryRate computes q-values for the IDs.
+- Finally, IDFilter selects only those IDs with a q-value of less than 1%.
+
+
+
+Extensions to this pipeline would be to do the annotation of the spectra with multiple search engines and combine the
+results afterwards, using the **ConsensusID** TOPP tool.
+
+The results may be exported using the **TextExporter** tool, for further analysis with different tools.
+
+## Quantitation of BSA runs
+
+The simple pipeline described in this section (`BSA_Quantitation.toppas`) can be used to quantify peptides that occur
+on different runs. The example dataset contains three different bovine serum albumin (BSA) runs. First, **FeatureFinderCentroided** is called since the dataset is centroided. The results of the feature finding are then annotated with (existing) identification results. For convenience, we provide these search results from OMSSA with peptides with an FDR of 5% in the BSA directory.
+
+
+
+Identifications are mapped to features by the **IDMapper**. The last step is performed by **FeatureLinkerUnlabeled** which links
+corresponding features. The results can be used to calculate ratios, for example. The data could also be exported to a
+text based format using the TextExporter for further processing (e.g., in Microsoft Excel).
+
+The results can be opened in TOPPView. The next figures show the results in 2D and 3D view, together with the feature
+intermediate results.
+
+```{tip}
+The intensities and retention times are slightly different between the runs. To correct for retention
+times shift, a map alignment could be done, either on the spectral data or on the feature data.
+```
+
+
+
+
+
+## Merger and Collect nodes
+
+The following example is trivial but demonstrates how merger and collector nodes can be used in a pipeline. Have a look
+at `merger_tutorial.toppas`:
+
+
+
+A Merger merges its incoming file lists, i.e., files of all incoming edges are appended into new
+lists (which have as many elements as the merger has incoming connections). All tools this merger has outgoing
+connections to are called with these merged lists as input files. All incoming connections should pass the same number
+of files (unless the corresponding preceding tool is in recycling mode).
+
+A collector node, on the other hand, waits for all rounds to finish before concatenating all files from all incoming
+connections into one single list. It then calls the next tool with this list of files as input. This will happen exactly
+once during the entire pipeline run.
+
+In order to track what is happening, open the example file and run it. When the pipeline execution has finished, have a
+look at all input and output files (e.g., select **Open in TOPPView** in the context menu of the input/output nodes). The
+input files are named `rt_1.mzML`, `rt_2.mzML`, ... and each contains a single spectrum with RT as indicated by the filename,
+which helps to understand which files have been merged together.
diff --git a/docs/tutorials/TOPPAS/general-introduction.md b/docs/tutorials/TOPPAS/general-introduction.md
new file mode 100644
index 00000000..afd02cf2
--- /dev/null
+++ b/docs/tutorials/TOPPAS/general-introduction.md
@@ -0,0 +1,20 @@
+General Introduction
+====================
+
+**TOPPAS** allows you to create, edit, open, save, and run TOPP workflows. Pipelines can be created conveniently in a
+GUI. The parameters of all involved tools can be edited within TOPPAS and are also saved as part of the pipeline
+definition in the `.toppas` file. Furthermore, **TOPPAS** interactively performs validity checks during the pipeline
+editing process and before execution (i.e., a dry run of the entire pipeline), in order to prevent the creation of
+invalid workflows. Once set up and saved, a workflow can also be run without the GUI using `ExecutePipeline -in .`
+
+The following figure shows a simple example pipeline that has just been created and executed successfully:
+
+
+
+To create a new TOPPAS file, do any of the following:
+
+- Open TOPPAS without providing any existing workflow - an empty workflow will be opened automatically.
+- In a running TOPPAS program, choose: **File** > **New**.
+- Create an empty file in your file browser (explorer) with the suffix `.toppas` and double-click it (on Windows systems
+ all `.toppas` files are associated with TOPPAS automatically during installation of OpenMS, on Linux, and macOS you
+ might need to manually associate the extension).
diff --git a/docs/tutorials/TOPPAS/user-interface.md b/docs/tutorials/TOPPAS/user-interface.md
new file mode 100644
index 00000000..d7956d03
--- /dev/null
+++ b/docs/tutorials/TOPPAS/user-interface.md
@@ -0,0 +1,166 @@
+User Interface
+=============
+
+## Introduction
+
+The following figure shows the **TOPPAS** main window and a pipeline which is just being created. The user has added
+some tools by drag and dropping them from the TOPP tool list on the left onto the central window. Additionally, the user
+has added nodes for input and output files.
+
+Next, the user has drawn some connections between the tools which determine the data flow of the pipeline. Connections
+an be drawn by first *deselecting* the desired source node (by clicking anywhere but not on another node) and then
+dragging the mouse from the source to the target node (if a *selected* node is dragged, it is moved on the canvas
+instead). When a connection is created and the source (the target) has more than one output (input) parameter, an
+input/output parameter mapping dialog shows up and lets the user select the output parameter of the source node and the
+input parameter of the target node for this data flow, shown here for the connection between PeptideIndexer and
+FalseDiscoveryRate. If the file types of the selected input and output parameters are not compatible with each other,
+**TOPPAS** will refuse to add the connection. It will also refuse to add a connection if it would create a cycle in the
+workflow, or if it just would not make sense, e.g. if its target is an input file node. The connection between the input
+file node (#1) and the OMSSAAdapter (#5) tool is painted yellow which indicates it is not ready yet, because no input
+files have been specified.
+
+
+
+The input/output mapping of connections can be changed at any time during the editing process by double-clicking an
+connections or by selecting **Edit I/O mapping** from the context menu which appears when a connection is right-clicked.
+All visible items (i.e. connections and the different kinds of nodes) have such a context menu. For a detailed list of
+the different menus and their entries, see [Menus](user-interface.md#menus).
+
+The following figure shows a possible next step: the user has double-clicked one of the tool nodes in order to configure
+its parameters. By default, the standard parameters are used for each tool. Again, this can also be done by selecting
+**Edit parameters** from the context menu of the tool.
+
+
+
+Once the pipeline has been set up, specify the input files before executing the pipeline. This is done by double-clicking
+an input node and selecting the desired files in the dialog that appears. Input nodes have a special mode named
+**recycling mode**, i.e., if the input node has fewer files than the following node has rounds (as it might have two incoming
+connections) then the files are recycled until all rounds are satisfied. This might be useful if one input node specifies a
+single database file (for a Search-Adapter like Mascot) and another input node has the actual MS2 experiments (which is
+usually more than one). Then the database input node would be set to `recycle` the database file, i.e. use it for every run of
+the MascotAdapter node. The input list can be recycled an arbitrary number of times, but the recycling has to be `complete`,
+i.e. the number of rounds of the downstream node have to be a multiple of the number of input files. Recycling mode can be
+activated by right-clicking the input node and selecting the according entry from the context menu. Finally, if you have input
+and output nodes at every end of your pipeline and all connections are green, select **Pipeline** > **Run** in the menu bar or
+just press F5.
+
+
+
+When asked for an output file directory `TOPPAS_out` as sub-directory, will be created. This directory will
+contain the output files. Specify the number of jobs TOPPAS is allowed to run in parallel. If a number greater than 1 is
+selected, TOPPAS will parallelize the pipeline execution in the following scenarios:
+
+- A tool has to process more than one file. In this case, multiple files can be processed in parallel.
+- The pipeline contains multiple branches that are independent of each other. In this case, multiple tools can run in
+ parallel.
+
+```{caution}
+Be careful with this setting, however, as some of the TOPP tools require large amounts of RAM (depending on the size of
+the dataset). Running too many parallel jobs on a machine with not enough memory will cause problems. Also, do not
+confuse this setting with the `threads` parameter of the individual TOPP tools: every TOPP tool has this parameter
+specifying the maximum number of threads the tool is allowed to use (although only a subset of the TOPP tools make use
+of this parameter, since there are tasks that cannot be computed in parallel). Be especially careful with combinations
+of both parameters! For a pipeline containing the `FeatureFinderCentroided`, as an example, and set its `threads`
+parameter to 8, and additionally set the number of parallel jobs in **TOPPAS** to 8, then you end up using 64 threads.
+```
+
+In addition to `TOPPAS_out`, a `TOPPAS_tmp` directory will be created in the OpenMS temp path (call the `OpenMSInfo`
+tool to see where exactly). It will contain all temporary files that are passed from tool to tool within the pipeline.
+Both folders contain further sub-directories which are named after the number in the top-left corner of the node they
+belong to (plus the name of the tool for temporary files). During pipeline execution, the status lights in the top-right
+corner of every tool show how many files have already been processed and the overall number of files to be processed by
+this tool. When the execution has finished, check the generated output files of every node quickly by selecting
+**Open files in TOPPView** or **Open containing folder** from the context menu (right click on the node).
+
+## Mouse and keyboard
+
+Using the mouse:
+
+- drag and drop tools from the TOPP tool list onto the workflow window (you can also double-click them instead)
+- select items (by clicking)
+- select multiple items (by holding down CTRL while clicking)
+- select multiple items (by holding down CTRL and dragging the mouse in order to "catch" items with a selection
+ rectangle)
+- move all selected items (by dragging one of them)
+- draw a new connection from one node to another (by dragging; source must be deselected first)
+- specify input files (by double-clicking an input node)
+- configure parameters of tools (by double-clicking a tool node)
+- specify the input/output mapping of connections (by double-clicking a connection)
+- translate the view (by dragging anywhere but on an item)
+- zoom in and out (using the mouse wheel)
+- make the context menu of an item appear (by right-clicking it)
+
+Using the keyboard:
+
+- delete all selected items (DEL or BACKSPACE)
+- zoom in and out (+ / -)
+- run the pipeline (F5)
+- open this tutorial (F1)
+
+Using the mouse + keyboard:
+
+- copy a node's parameters to another node (only parameters with identical names will be copied, e.g.,
+ `fixed_modifications`) (CTRL while creating an edge) The edge will be colored as dark magenta to indicate parameter
+ copying.
+
+## Menus
+
+### Menu bar
+
+In the **File** menu:
+
+- create a new, empty workflow (**New**)
+- open an existing one (**Open**)
+- open an example file (**Open example file**)
+- include an existing workflow to the current workflow (**Include**)
+- visit the online workflow repository (**Online repository**)
+- save a workflow (**Save**/**Save as**)
+- export the workflow as image (**Export as image**)
+- refresh the parameter definitions of all tools contained in the workflow (**Refresh parameters**)
+- close the current window (**Close**)
+- load and save TOPPAS resource files (`.trf`) (**Load**/**Save OPPAS resource file**)
+
+In the **Pipeline** menu:
+
+- run a pipeline (**Run**)
+- abort a currently running pipeline (**Abort**)
+
+In the **Windows** menu:
+
+- make the TOPP tool list window on the left, the description window on the right, and the log message at the bottom
+ (in)visible.
+
+In the **Help** menu:
+
+- go to the OpenMS website (**OpenMS website**)
+- open this tutorial (**TOPPAS tutorial**)
+
+### Context menus
+
+In the context menu of an `input node`:
+
+- specify the input files
+- open the specified files in TOPPView
+- open the input files' folder in the window manager (explorer)
+- toggle the **recycling** mode
+- copy, cut, and remove the node
+
+In the context menu of a `tool`:
+
+- configure the parameters of the tool
+- resume the pipeline at this node
+- open its temporary output files in TOPPView
+- open the temporary output folder in the file manager (explorer)
+- toggle the **recycling** mode
+- copy, cut, and remove the node
+
+In the context menu of a `Merger` or `Collector`:
+
+- toggle the **recycling** mode
+- copy, cut, and remove the node
+
+In the context menu of an `output node`:
+
+- open the output files in TOPPView
+- open the output files folder in the window manager (explorer)
+- copy, cut, and remove the node
diff --git a/requirements.txt b/requirements.txt
new file mode 100644
index 00000000..06400885
--- /dev/null
+++ b/requirements.txt
@@ -0,0 +1,9 @@
+sphinx
+readthedocs-sphinx-search
+furo
+sphinx-copybutton
+myst-parser
+sphinx-notfound-page
+sphinxcontrib-images
+sphinx-inline-tabs
+pygments
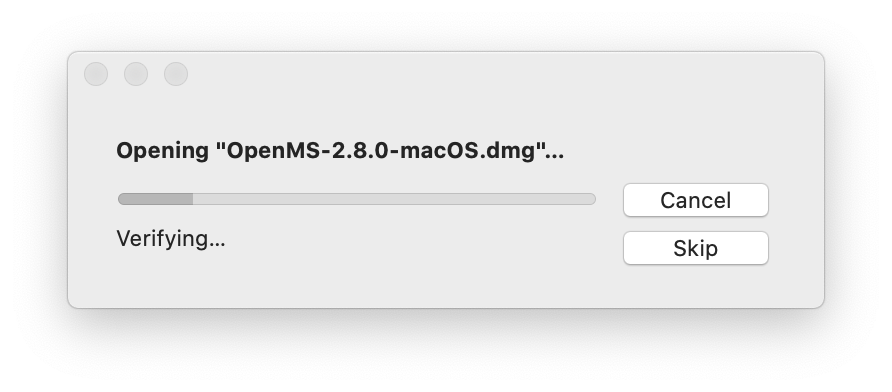 +
+3. Verify the download.
+
+
+
+3. Verify the download.
+
+ +
+4. Agree the license agreements.
+
+
+
+4. Agree the license agreements.
+
+ +
+5. Drag openms to applications.
+
+
+
+5. Drag openms to applications.
+
+ +
+6. It will start copying to applications.
+
+
+
+6. It will start copying to applications.
+
+ +
+
+
+ +
+To use {term}`TOPP` as regular app in the shell, add the following lines to the `~/.profile` file.
+
+```bash
+export OPENMS_TOPP_PATH=
+
+To use {term}`TOPP` as regular app in the shell, add the following lines to the `~/.profile` file.
+
+```bash
+export OPENMS_TOPP_PATH=