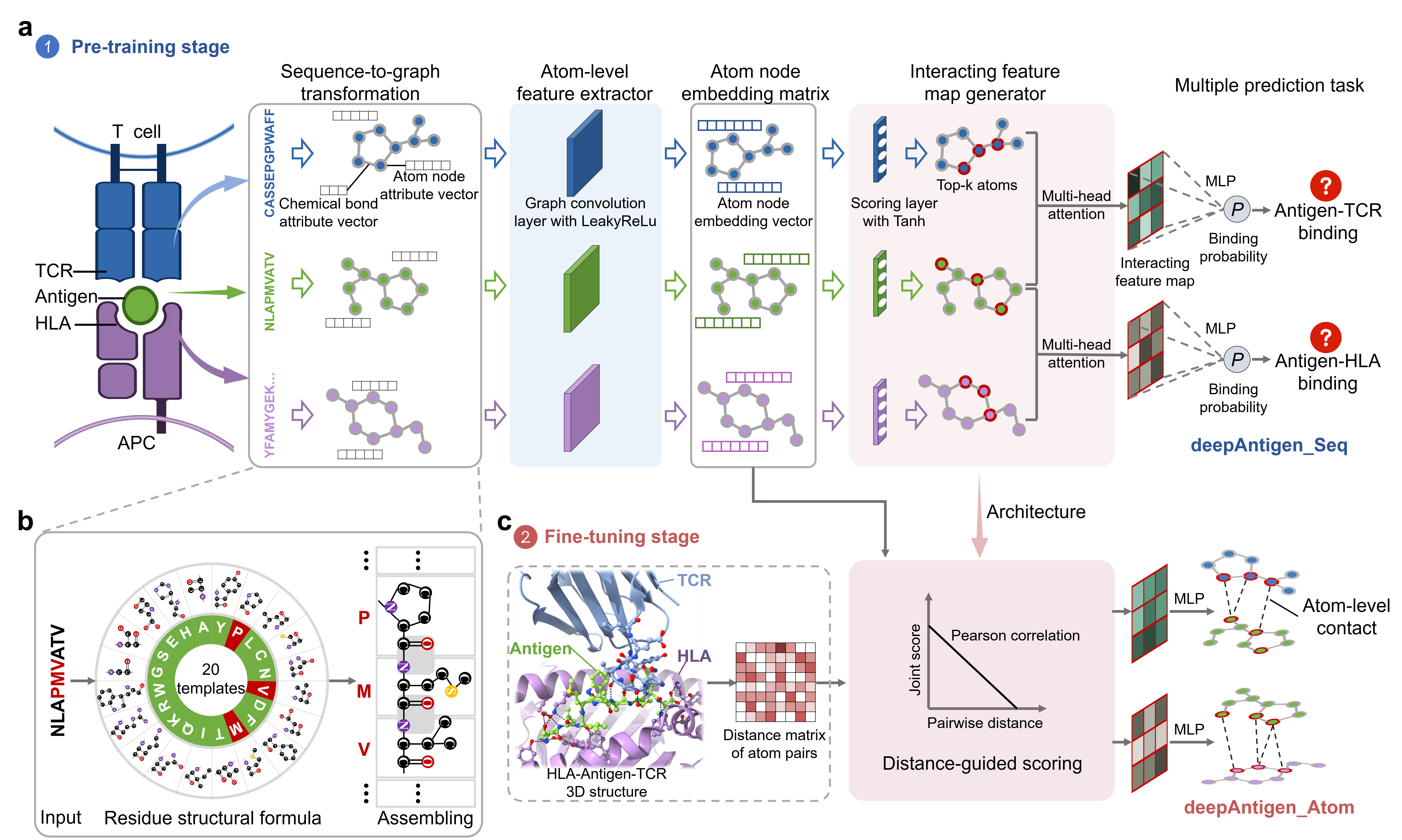
To install deepAntigen, make sure you have installed PyTorch and PyTorch Geometric. If you need more details on the dependences, look at the environment.yml file.
- set up conda environment for deepAntigen
conda create -n deepAntigen-env python=3.8- install deepAntigen from shell
pip install deepAntigen
pip install torch==1.9.0+cu111 -f https://download.pytorch.org/whl/torch_stable.html
pip install torch-cluster==1.5.9 torch-scatter==2.0.7 torch-sparse==0.6.12 torch-spline-conv==1.2.1 -f https://data.pyg.org/whl/torch-1.9.0%2Bcu111.html
pip install torch-geometric==2.4.0Using deepAntigen to achieve different tasks, please import corresponding module to your jupyter notebooks or scripts.
If you want to predict antigen-HLAI binding at the sequence level,
from deepantigen.antigenHLAI import run_antigenHLAI_seqIf you want to predict atom-level contact between antigen and HLAI,
from deepantigen.antigenHLAI import run_antigenHLAI_atomIf you want to predict antigen-HLAII binding at the sequence level,
from deepantigen.antigenHLAII import run_antigenHLAII_seqIf you want to predict atom-level contact between antigen and HLAII,
from deepantigen.antigenHLAII import run_antigenHLAII_atomIf you want to predict antigen-TCR binding at the sequence level,
from deepantigen.antigenTCR import run_antigenTCR_seqIf you want to predict atom-level contact between antigen and TCR,
from deepantigen.antigenTCR import run_antigenTCR_atomFor sequence-level prediction, please prepare your antigen-HLAI data and place them in a .csv file format similar to the test_antigenHLAI/sequence/test.csv provided. The column 'label' is optional.
df = run_antigenHLAI_seq.Inference(path)The returned DataFrame, df, is prediction results of deepAntigen, which includes the binding probability for each antigen-HLAI pair.
For atom-level prediction, please prepare your antigen-HLAI data and place them in a .csv file format similar to the test_antigenHLAI/crystal_structure/sample.csv provided.
peptide_atoms, HLAI_atoms, contact_maps = run_antigenHLAI_atom.Inference(path)The returned three lists correspond top-k atoms of the peptide, top-k atoms of the HLAI and atom-level contact probability. Each element in peptide_atoms orHLAI_atoms is a list with length of k. Each element in contact_maps is a k*k DataFrame.
If you want to train deepAntigen with your own antigen-HLAI binding data, please reference the detailed Documentaion about deepAntigen.
For sequence-level prediction, please prepare your antigen-HLAII data and place them in a .csv file format similar to the test_antigenHLAII/sequence/test.csv provided. The column 'label' is optional.
df = run_antigenHLAII_seq.Inference(path)The returned DataFrame, df, is prediction results of deepAntigen, which includes the binding probability for each antigen-HLAII pair.
For atom-level prediction, please prepare your antigen-HLAII data and place them in a .csv file format similar to the test_antigenHLAII/crystal_structure/sample.csv provided.
peptide_atoms, HLAII_atoms, contact_maps = run_antigenHLAII_atom.Inference(path)The returned three lists correspond top-k atoms of the peptide, top-k atoms of the HLAII and atom-level contact probability. Each element in peptide_atoms or HLAII_atoms is a list with length of k. Each element in contact_maps is a k*k DataFrame.
If you want to train deepAntigen with your own antigen-HLAII binding data, please reference the detailed Documentaion about deepAntigen.
For sequence-level prediction, please prepare your antigen-TCR data and place them in a .csv file format similar to the test_antigenTCR/sequence/test.csv provided. The column 'label' is optional.
df = run_antigenTCR_seq.Inference(path)The returned DataFrame, df, is prediction results of deepAntigen, which includes the binding probability for each antigen-TCR pair.
For atom-level prediction, please prepare your antigen-TCR data and place them in a .csv file format similar to the test_antigenTCR/crystal_structure/sample.csv provided.
peptide_atoms, TCR_atoms, contact_maps = run_antigenTCR_atom.Inference(path)The returned three lists correspond top-k atoms of the peptide, top-k atoms of the TCR and atom-level contact probability. Each element in peptide_atoms or TCR_atoms is a list with length of k. Each element in contact_maps is a k*k DataFrame.
If you want to train deepAntigen with your own antigen-TCR binding data, please reference the detailed Documentaion about deepAntigen.
See detailed documentation and examples at https://deepAntigen.readthedocs.io/en/latest/index.html.
Feel free to submit an issue or contact us at quejinhao2021@163.com for problems about the package.