Conversation
 richardjgowers
left a comment
richardjgowers
left a comment
There was a problem hiding this comment.
Would it be possible to have a smarts keyword instead of forcing one type or another of selection? Eg "smarts [#7] and not resname ASP"
|
Since some SMARTS have parenthesis in them I'd need to find a way to bypass the "split original selection string on spaces and parenthesis" thing, but should be doable |
|
Done! |
|
This is pretty awesome btw :) |
| if not pattern: | ||
| raise ValueError("{!r} is not a valid SMARTS query".format( | ||
| self.pattern)) | ||
| mol = group.convert_to("RDKIT") |
There was a problem hiding this comment.
Might be nice to try and cache this in Topology.
There was a problem hiding this comment.
The mol (topology, not coordinates) is already cached in the converter so that we don't need to rebuild it for every frame of a trajectory:
https://github.com/cbouy/mdanalysis/blob/rdkit-converter/package/MDAnalysis/coordinates/RDKit.py#L253-L264
package/MDAnalysis/core/selection.py
Outdated
| mol = group.convert_to("RDKIT") | ||
| matches = mol.GetSubstructMatches(pattern, useChirality=True) | ||
| indices = [idx for match in matches for idx in match] | ||
| indices = [mol.GetAtomWithIdx(i).GetIntProp("_MDAnalysis_index") |
There was a problem hiding this comment.
Can we not predict what idx RDKit will assign? Ie do the atoms sometimes get reordered?
There was a problem hiding this comment.
They get reordered when using reactions to "standardize" some functional groups like nitro, sulfone...etc into the correct form. I could also force reordering atoms at the end of the conversion but that requires building a new molecule again I think.
| token = 'smarts' | ||
|
|
||
| def __init__(self, parser, tokens): | ||
| pattern = [] |
There was a problem hiding this comment.
Is it simpler to enforce no white space allowed in the pattern?
There was a problem hiding this comment.
White spaces are added automatically by the parser around parentheses:
https://github.com/cbouy/mdanalysis/blob/rdkit-converter/package/MDAnalysis/core/selection.py#L1182
|
That is a super cool feature! I did not read the code so maybe it is obvious and super well documented, but I have 2 questions:
|
It raises an ImportError with a message that you need RDKit to use the SMARTS selection
You get a flat and "uniqued" AtomGroup with all atoms matching the query. For example, with the molecule RDKit returns a tuple of matches for the query, i.e. if you query There's a limit of 1000 matches (the default in RDKit). Maybe it could be useful to add a kwarg to |
For API consistency's sake, it would be better to have an other method to do this. I think it would be very useful, but it is clearly for later. |
|
I agree that it could get confusing, a separate method is probably better for ag in u.smarts_matches("C-C"):
print(ag.indices) |
richardjgowers
left a comment
There was a problem hiding this comment.
Code is fine, quick doc question
 IAlibay
left a comment
IAlibay
left a comment
There was a problem hiding this comment.
Couple of things, but otherwise lgtm :)
 fiona-naughton
left a comment
fiona-naughton
left a comment
There was a problem hiding this comment.
Just one comment from me, otherwise looking good!
IAlibay
left a comment
There was a problem hiding this comment.
Assuming CI returns green lgtm!
pinging @fiona-naughton and @richardjgowers for re-review, we can probably get this merged today/tomorrow.
Codecov Report
@@ Coverage Diff @@
## develop #2883 +/- ##
===========================================
+ Coverage 92.91% 93.01% +0.10%
===========================================
Files 187 187
Lines 24591 24963 +372
Branches 3185 3263 +78
===========================================
+ Hits 22848 23219 +371
+ Misses 1697 1696 -1
- Partials 46 48 +2
Continue to review full report at Codecov.
|

Towards MDAnalysis#2468 ## Work done in this PR Adds a new SMARTS based selection which uses the RDKit converter.
Part of #2468
Depends on #2775
Changes made in this Pull Request:
u.select_atoms("[C;R;!a]", smarts=True)An example with Proline:
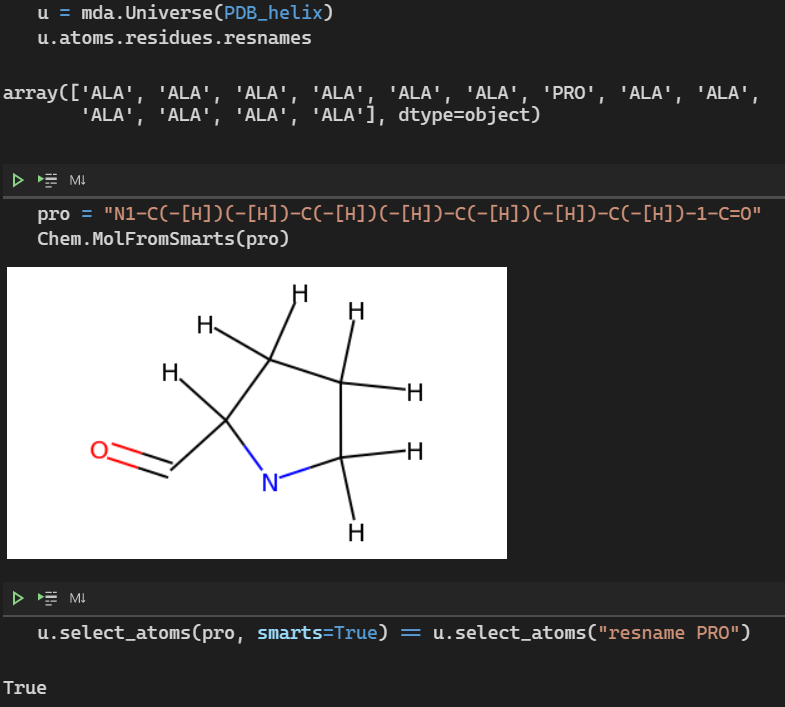
PR Checklist
@richardjgowers @IAlibay @fiona-naughton