![]() tricky problems, but smart solutions for Computational Chemistry
tricky problems, but smart solutions for Computational Chemistry
These are some of the main codes I have used to solve common problems I've come to face throughout my PhD. I hope they will be useful to you, too! :)
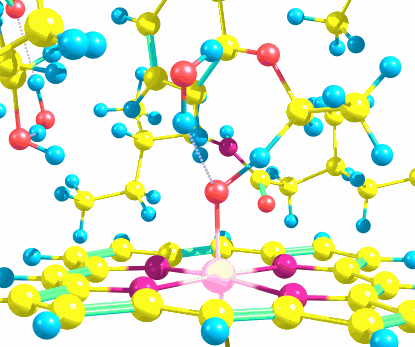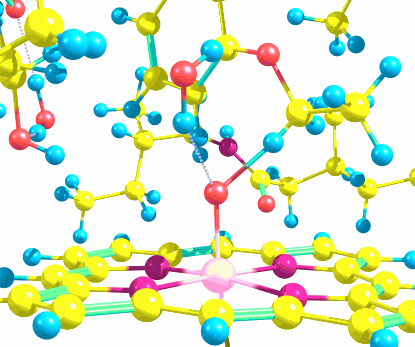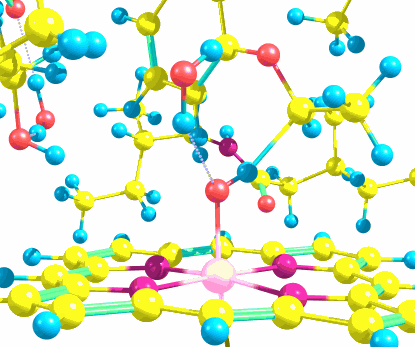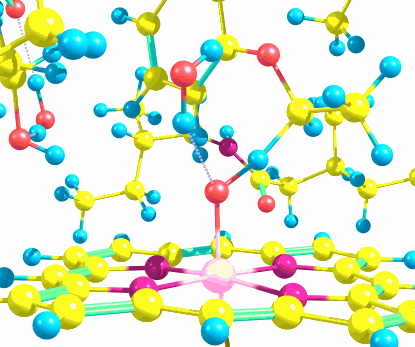
Use this script to extract the energies of your DFT system, as well as charges and spin distribuitions of all components of it, as outputted by Gaussian. This script makes the data extraction much faster without the need to open the usually too large Gaussian output file and creates an easy-to-read text file with all relevant information for chemical reaction modelling regarding that specific structure, be it a reactant, an intermediate, a product or even a transition state. To that goal, you will have to adapt the code to your system's specific atomic numbering, as described in the comments within the script.
Use this script to create an energy landscape of a chemical reaction pathway, starting from two output files of the above 'extract.py' script. This script outputs the energy differences between two structures, the first of which should be the reactant/reference starting structure. You can use it multiple times to assemble your specific reaction energy landscape, but just remember to keep the same reference structure in each iteration.
Use this script to get a list of the protonation states of all ASP, GLU, and HIS (HSP, HSE, or HSD) in your protein. It reads the PDB file outputted by H++ webserver and lists all protonation states of titratable residues, and this information can then be used in the creation of the topology file. This will save you a lot of time that you would otherwise spend inspecting visually the protein structure outputted by H++.
Use this script to convert all charmm-named atoms into amber-named ones. This will be useful when working with ParmEd.
Use this script to obtain the normalized average of a molecular property extracted from an MD trajectory (rmsd, distance, angle, for instance). The input files for this program are a series of output files obtained from cpptraj-based analysis of MD simulations, like the ones given off by the modules lie, rmsd, distance, angleinfo and dihedral. The result is a list of normalized average values for each property described in each input file.
Use this script to correct large files that have ***** in the place where there should be a number. For instance, in the bottom of large files, you can usually find ***** when you run out of decimal places. Please see the comment lines inside the script for a better sense of how it works.
Use this script to extract all frames from a molecular dynamics trajectory and convert each one into PDB files that are CAVER-friendly, that is, easy to be read by the cavity/tunnel-finding software CAVER.
Enjoy!