![]()
![]()

Note: this package is in early stages of development.
scLoop is a library to identify statistically significant loops in single-cell RNA-seq data.
import scloop as scl
scl.pp.prepare_adata(adata, downsample=True, n_downsample=500)
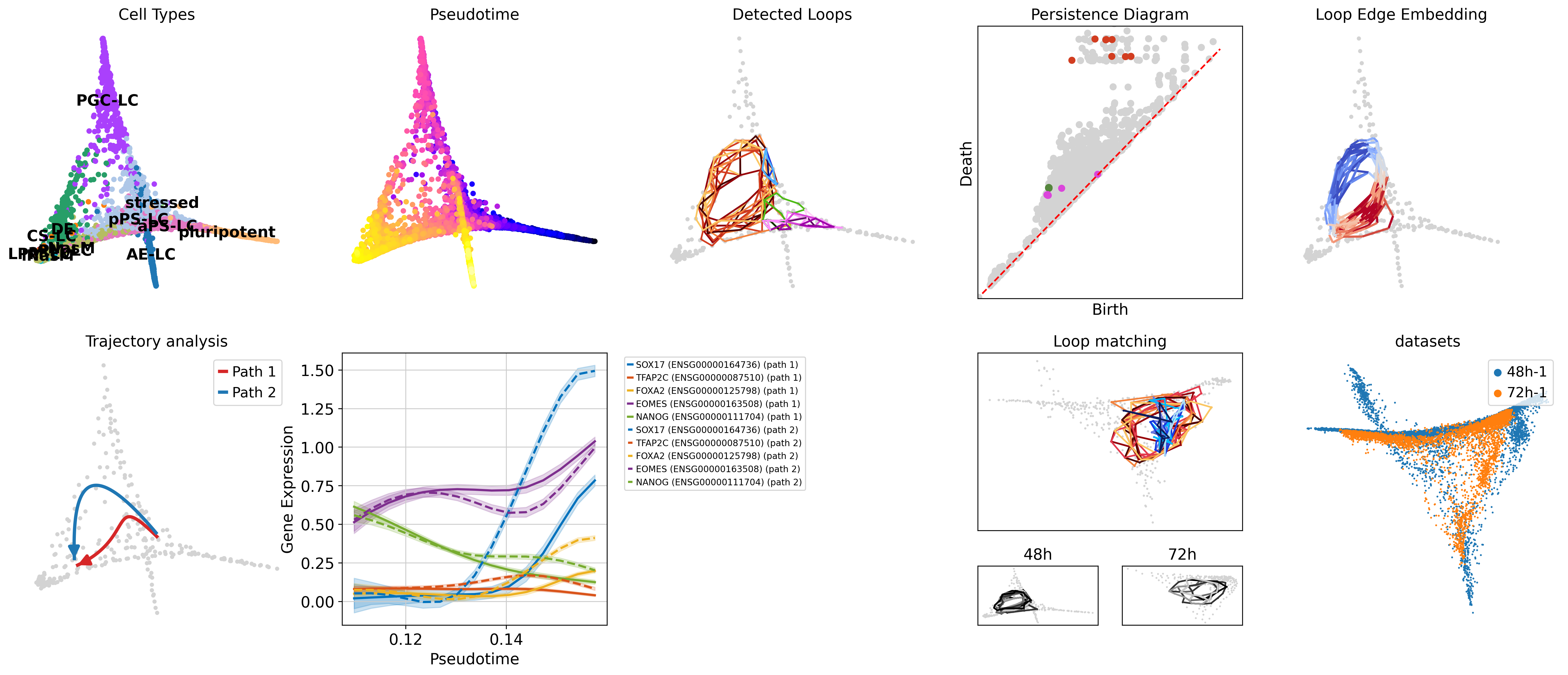
scl.tl.find_loops(adata, ...)
scl.tl.analyze_loops(adata, ...)
scl.tl.match_loops([adata1, adata2, ...], ...)make buildor
make rebuildmake syncor
make full-syncto prevent partial compilation of some modules
Note: this package is in early stages of development. The current build will have issues.
pip install scloopsrc/scloop/
├── analyzing
│ ├── bootstrap.py
│ ├── feature_selection.py
│ ├── gene_trend.py
│ ├── hodge.py
│ ├── __init__.py
│ └── stats.py
├── benchmarking
│ └── __init__.py
├── computing
│ ├── boundary.py
│ ├── hodge_decomposition.py
│ ├── hodge.py
│ ├── homology.py
│ ├── __init__.py
│ ├── loops.py
│ └── matching.py
├── data
│ ├── analysis_containers.py
│ ├── base_components.py
│ ├── boundary.py
│ ├── constants.py
│ ├── containers.py
│ ├── __init__.py
│ ├── metadata.py
│ ├── ripser.cpp
│ ├── ripser.hpp
│ ├── ripser_lib.cpp
│ ├── ripser_lib.pyx
│ ├── types.py
│ └── utils.py
├── __init__.py
├── matching
│ ├── cross_dataset.py
│ ├── data_modules.py
│ ├── __init__.py
│ ├── mlp.py
│ └── nf.py
├── plotting
│ ├── _cross_match.py
│ ├── _hodge.py
│ ├── _homology.py
│ ├── __init__.py
│ └── _utils.py
├── preprocessing
│ ├── delve
│ │ ├── delve.py
│ │ ├── __init__.py
│ │ └── kh.py
│ ├── downsample.py
│ ├── __init__.py
│ └── prepare.py
├── py.typed
├── tools
│ ├── _cross_match.py
│ ├── __init__.py
│ └── _loops.py
└── utils
├── distance_metrics
│ ├── discrete-frechet-distance
│ ├── frechet.cpp
│ ├── frechet_py.py
│ ├── frechet.pyx
│ └── __init__.py
├── __init__.py
├── linear_algebra_gf2
│ ├── gf2_toolkit_lib.pyx
│ ├── __init__.py
│ ├── m4ri_lib.c
│ └── m4ri_lib.pyx
├── logging.py
└── pvalues.py