Added fitting transformations#1991
Conversation
Codecov Report
@@ Coverage Diff @@
## develop #1991 +/- ##
==========================================
+ Coverage 89.66% 89.7% +0.03%
==========================================
Files 172 173 +1
Lines 21421 21493 +72
Branches 2792 2799 +7
==========================================
+ Hits 19208 19280 +72
Misses 1616 1616
Partials 597 597
Continue to review full report at Codecov.
|

|
I added a simpler form of alignto, but modified to remove rotation and translation on a given plane. I added a test to the output coordinates to check if the code is working. I'll add more extensive tests later |
|
The calculations were wrong in the This While correcting the errors in this function I realised I made some mistakes in PR #1937. I'll have to open an issue to correct those. Now, I face an issue. When I use atom masses as weights for If alignto stays the same, how do I test this "similarity" in my transformations? |
 jbarnoud
left a comment
jbarnoud
left a comment
There was a problem hiding this comment.
I probably missed many things. The quality of this PR is much better than the previous ones. I am ery happy with the progress I see :)
I am not sure I understand. Could you elaborate? |
| return wrapped | ||
|
|
||
|
|
||
| def fit_rot_trans(ag, reference, plane=None, weights=None): |
There was a problem hiding this comment.
I see what makes this different from alignto but it seems to me that alignto here is just a special case of this function. Why not have it all in one instead of two confusingly similar functions?
|
Also, for all your PRs, have a look at the coverage report. |
|
@jbarnoud @kain88-de Sorry for the delay. The way I was calculating the plane rotation and translation fittings was not correct. I was trying to find an alternative to finding the euler angles of the rmsd fitting rotation matrix. Now, I just need some suggestions for testing this plane fitting feature. |
|
The plane fitting is the same as transxy in trjconv, right? You can use the DCD system from the test data files, translate it in the 3 directions and make sure that 2 coordinates are the same as before once you apply the fitting. You can parametrize the test to work on the 3 planes separately. Le 25 juil. 2018 12:10 AM, Davide Cruz <notifications@github.com> a écrit :@jbarnoud @kain88-de Sorry for the delay. The way I was calculating the plane rotation and translation fittings was not correct. I was trying to find an alternative to finding the euler angles of the rmsd fitting rotation matrix.
One idea would be to perform a 2D rmsd fitting. I decided then to commit what I had and then research on how to write that fitting.
However, when I was testing my code against the output of trjconv -fit rotxy+transxy on the trajectory of a membrane peptide and, to my surprise, they were equal ( .07A RMSD)!
Now, I just need some suggestions for testing this plane fitting feature.
—You are receiving this because you were mentioned.Reply to this email directly, view it on GitHub, or mute the thread.
|
|
@jbarnoud Actually, rotxy+transxy does'n work like that. When fitting to the xy plane, only the z coordinates are preserved. Thus the group that you choose to fit will not rotate on the z axis (so you rotate it on Z every frame so it becomes as close as possible to the reference), but will translate on the z axis - but not in the x and y axis. This makes this fitting useful when, for example, you want to see molecule insertion in membranes, or keep the molecule as static as possible while keeping the membrane horizontal. |
|
I thought you were talking about the transxy without rotation.If you build artificial systems, you can apply the transformation, then apply the part of the rotation that is missing to get a full fit. The transformed coordinates should be the same as the reference. If the transformation you are testing rotate in an unexpected way, there should be a mismatch. Le 26 juil. 2018 6:47 PM, Davide Cruz <notifications@github.com> a écrit :@jbarnoud Actually, rotxy+transxy does'n work like that. When fitting to the xy plane, only the z coordinates are preserved. Thus the group that you choose to fit will not rotate on the z axis (so you rotate it on every frame so it becomes as close as possible to the reference), but will translate on the z axis - but not in the x and y axis. This makes this fitting useful when, for example, you want to see molecule insertion in membranes, or keep the molecule as static as possible while keeping the membrane horizontal.
—You are receiving this because you were mentioned.Reply to this email directly, view it on GitHub, or mute the thread.
|
|
@davidercruz (and @jbarnoud ?) could you please summarize what needs to be done to get this PR done? I am not looking for a promise that this gets done over the next weekend/holidays but something that will help to get the work done. This is an open source project so I understand that everyone is short on time and priorities shift but it would be a real shame if this didn't make it into MDAnalysis. It is always possible that someone else might find the energy to finish it (e.g., @ianmkenney in my group has recently been playing with the OTFT) but in this case one should make it easy for others to pick up the work, especially if one has not been able to finish things. |
|
I am almost sure it is ready to go. I just didn't find the time to try it on a real system. On 14 Dec 2018 21:39, Oliver Beckstein <notifications@github.com> wrote:@davidercruz (and @jbarnoud ?) could you please summarize what needs to be done to get this PR done?
I am not looking for a promise that this gets done over the next weekend/holidays but something that will help to get the work done. This is an open source project so I understand that everyone is short on time and priorities shift but it would be a real shame if this didn't make it into MDAnalysis. It is always possible that someone else might find the energy to finish it (e.g., @ianmkenney in my group has recently been playing with the OTFT) but in this case one should make it easy for others to pick up the work, especially if one has not been able to finish things.
—You are receiving this because you were mentioned.Reply to this email directly, view it on GitHub, or mute the thread.
|
|
@jbarnoud @richardjgowers @davidercruz any chance that this gets into 0.20.0 #2240 (as we had promised in the 0.19.x blog post)? |
- fix #13 - needs MDAnalysisData >= 0.7.0 for the "membrane peptide" dataset - needs MDAnalysis >= 0.20.0 for the transformations (currently PRs MDAnalysis/mdanalysis#2038 , MDAnalysis/mdanalysis#1991 , and MDAnalysis/mdanalysis#1973 ) - updated text in notebook - adde a few more empty lines for clarity
 tylerjereddy
left a comment
tylerjereddy
left a comment
There was a problem hiding this comment.
TestEncore.test_triangular_matrix was failing in some CI entries--do we need some skips there? I know we've had issues with encore test fragility in the past & doesn't seem to be related to changes here.
I ran coverage and full test suite on this branch locally & it seemed okay. Pretty unlikely to break any old code at first glance, since it is effectively a new module & no old tests modified.
We should mark as "resolved" all reviewer comments that have been addressed, so that it is clearer which are outstanding--I'll try to start on that now.
|
Oh right, we patched a CVE vulnerability in NumPy upstream related to pickling and |
|
I've made a note to return to this PR Thursday--hopefully by then the CVE-related test failure is resolved / merged & this PR can be presented with CI all green / coverage solid. Looks like we almost have an approving review from @jbarnoud, so shouldn't be that hard to push this over the line now. |
|
Thank you @tylerjereddy |
- fix #13 - needs MDAnalysisData >= 0.7.0 for the "membrane peptide" dataset - needs MDAnalysis >= 0.20.0 for the transformations (currently PRs MDAnalysis/mdanalysis#2038 , MDAnalysis/mdanalysis#1991 , and MDAnalysis/mdanalysis#1973 ) - updated text in notebook - adde a few more empty lines for clarity
231a2c5 to
ba26315
Compare
|
Rebased on latest develop branch--hopefully this gets us to green CI with a cov report for reviewers to see. |
ba26315 to
20b6594
Compare
|
CI was all green, but I've tried to bump test coverage up to 100% on the patch diff. Marked one more reviewer comment as resolved. |
|
Our CI is definitely prone to issues---looks like CI helpers is frequently stalling in Travis. Whoever is / was in favor of CI helpers may want to work with them to smooth it out. |
|
@richardjgowers Does the bottom right plot above make intuitive sense to you? |
|
@tylerjereddy I'm not 100% on how this all works, but looks like a bug to me, I'd think that the center should still remain constant, just it's defined as COM not COG. |
I'm not so sure--I've been thinking about this and produced a plot that tracks the center of mass and that is indeed fixed as you can see below for the same case as bottom right centroid-tracking plot above. So, this means that
|
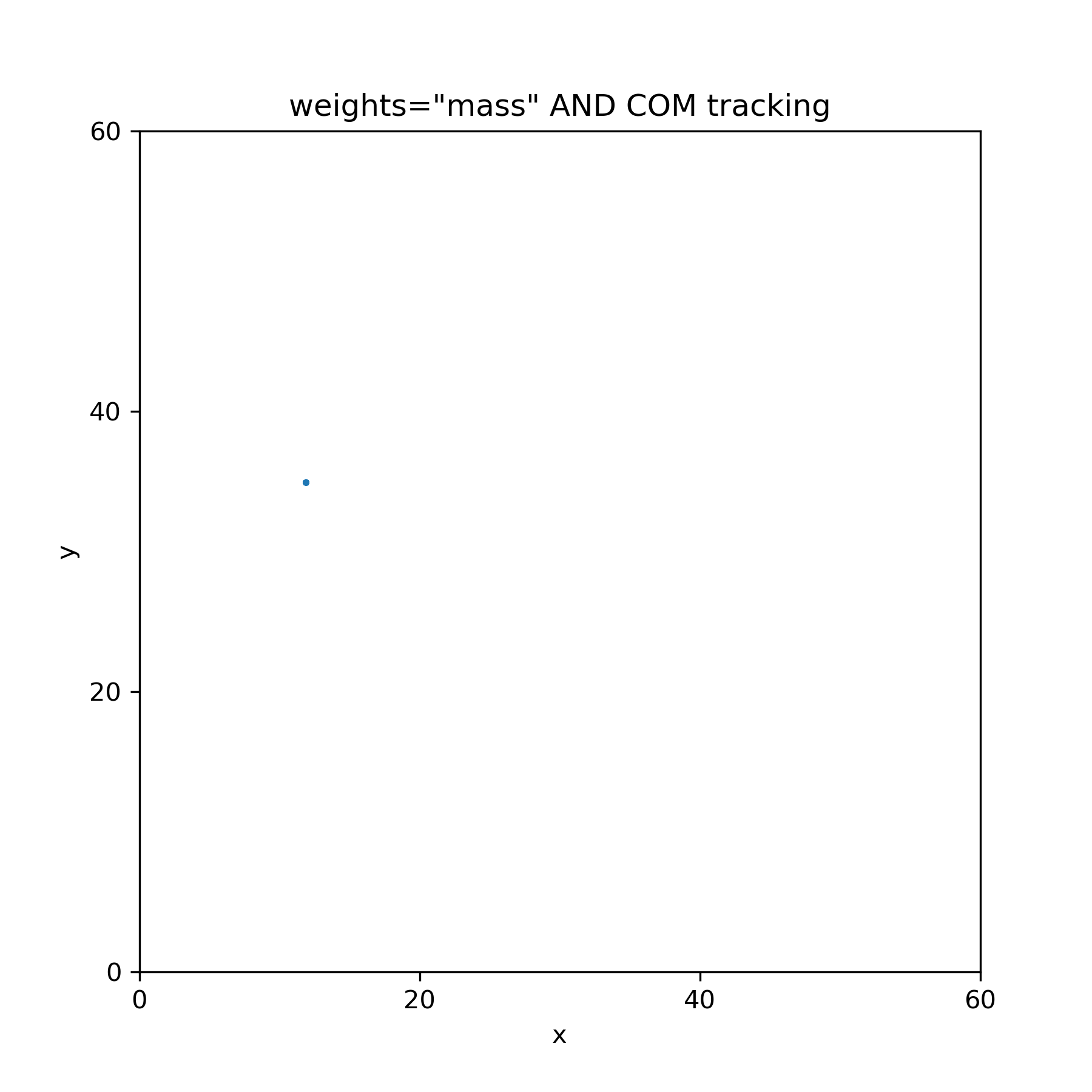
|
If we can agree that is reasonable then I'll move on to evaluating the other fitting function. |
Stacking should be faster because loading data from disk is typically 5-10x more costly than the actual calculation, at least for "simple" (or highly optimized) things such as RMSD. If you can load once and calculate twice, you can probably get almost 2x overall speed-ups. |
The only time when COG is always guaranteed to be the same as COM is when all masses are identical. Otherwise they should be different (in general – I think with an inversion symmetry they will still be identical but that seems far fetched here and easily destroyed by thermal motions).
I agree that this is reasonable. Thank you for the detailed evaluation! |
tylerjereddy
left a comment
There was a problem hiding this comment.
Expanding the figure panel to include the second fitting function in this PR suggests that they can produce very similar results, which is a good sign:
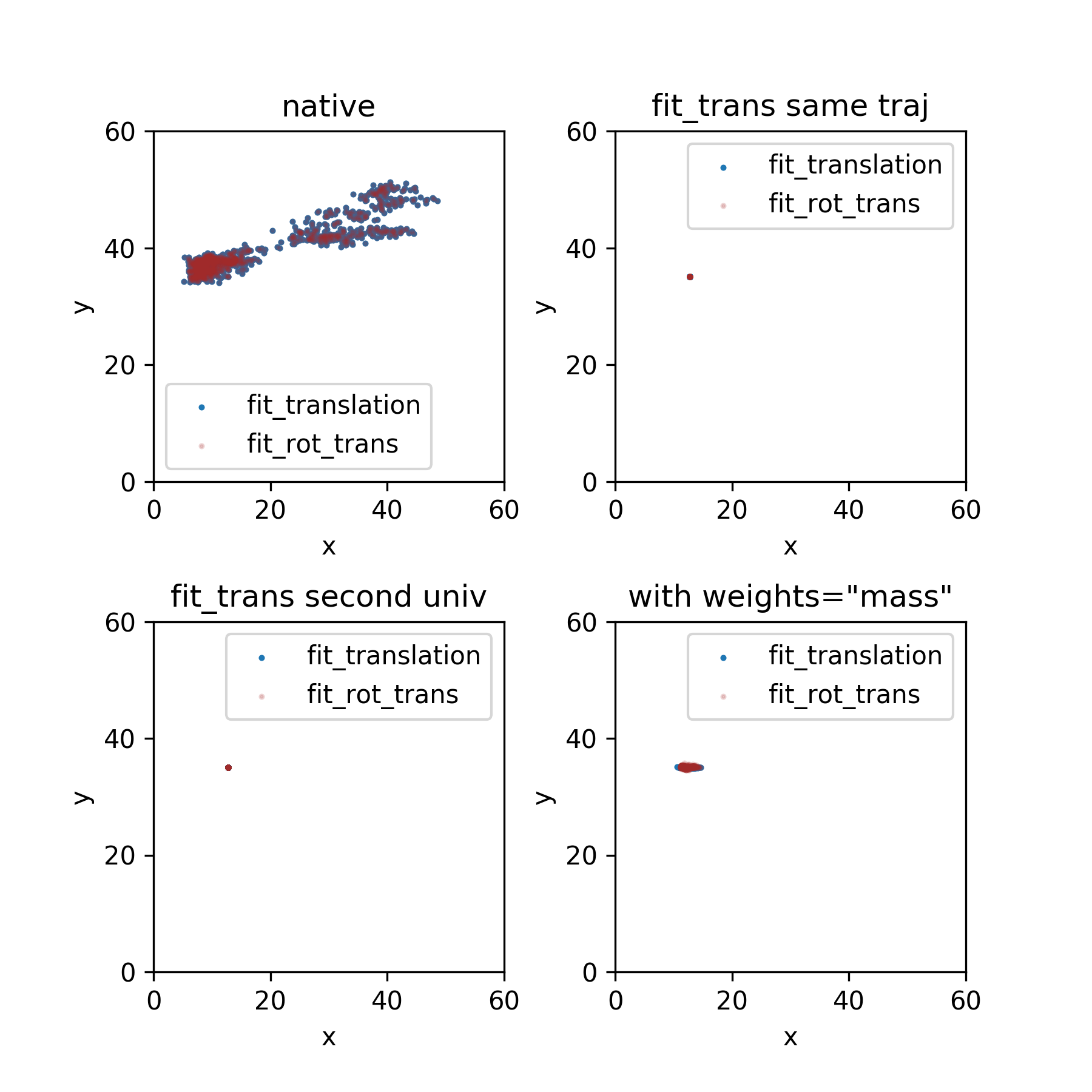
However, my first draft comparison of the rmsd-to-reference profiles is a little strange on the magnitude side, albeit still sensibly favoring the the RMSD-based approach. Nonetheless, I suspect these numbers are a little off--perhaps something is off in my test code:
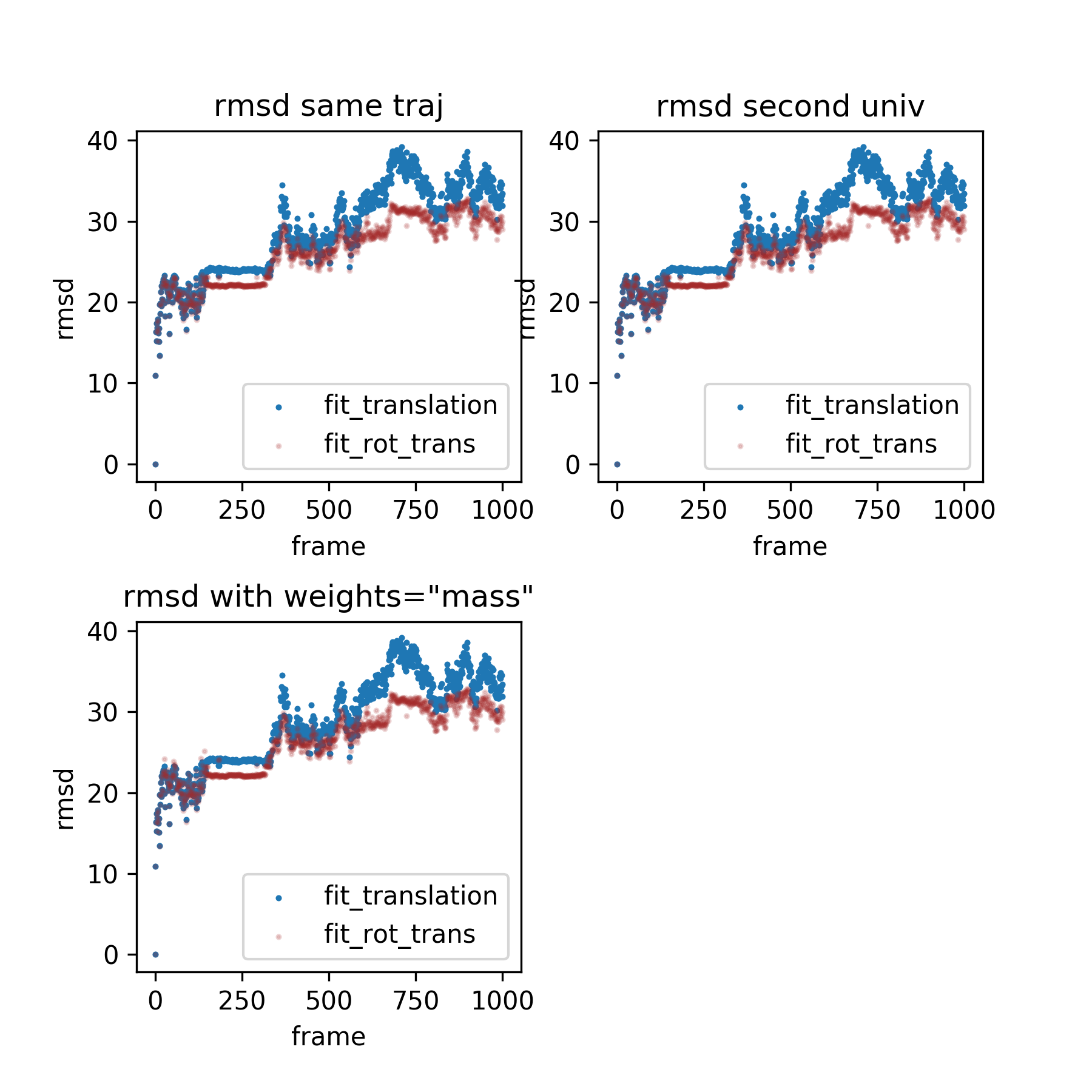
I've assumed that using MDAnalysis.analysis.rms.rmsd(ag.positions, copy_of_ref_positions) would suffice throughout.
Once we sort through this RMSD thing, I think I can just push in my minor changes & that's about it.
|
@tylerjereddy the RMSD is so high because the peptide in the trajectory is broken across the box. I just visualized it. If this is all then I think this looks sensible. |
tylerjereddy
left a comment
There was a problem hiding this comment.
Ok, I've pushed in revisions to address my own minor doc refinements & am now marking this as approved.
There's just one unresolved reviewer comment left, and while Max's concern that alignto has some API overlap is solid, I do suggest we accept the minor blemish & move on / forward, given our constraints on reviewer bandwidth and so on.
|
@orbeckst Thanks, I often work on WSL in the evenings, so it is a little more annoying to try doing visualizations from command line. |
|
Feel free to rebase if you feel the need, but as long as there are no merge conflicts I suspect most of our CI just does merges into develop anyway. |
|
Looks like one of the Travis entries has a setup issue of some sort. Maybe a second version of NumPy is getting picked up or something like that. |
|
The problem is that conda installs a too old scipy version even though we require >1.0. Later, when the And a little further below: |
|
Oy, thanks. That should probably be simplified & ideally hard fail earlier on if there's a problem, I suspect. |
|
@zemanj could you please raise an issue with your problem diagnosis #1991 (comment) ? Many thanks!! |
* test_fit.py now uses proper __future__ imports, which should alleviate code style / linting issues for old style division * test_fit.py adjusted to bump coverage of fit.py to 100 %
* just minor DOC adjustments noticed in a final review
ad5ec8f to
df3684c
Compare
|
Rebased on latest develop & force pushed -- hopefully CI is finally green |
|
CI is all green -- in it goes. The original PR description includes a checkmark for CHANGELOG entry, but I don't see that. It can always be done in a follow-up. Thanks @davidercruz & others. |
|
Thank you all for the hard work getting it in!! |
Changes made in this Pull Request:
alignto, from theMDAnalysis.analysis.alignmodule, as a way to remove rotations and translations of a given AtomGroup in a trajectoryTODO in this PR:
PR Checklist