Make H5MD serialize with pickle #2894
+151
−28
There are no files selected for viewing
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
|
|
@@ -31,7 +31,7 @@ | |
| interface of the HDF5 library to improve parallel reads and writes. | ||
|
|
||
| The HDF5 library and `h5py`_ must be installed; otherwise, H5MD files | ||
| cannot be read by MDAnalysis. If `h5py`_ is not installed, a | ||
| :exc:`RuntimeError` is raised. | ||
|
|
||
| Units | ||
|
|
@@ -94,10 +94,10 @@ | |
|
|
||
| Building parallel h5py and HDF5 on Linux | ||
| ^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^^ | ||
| Building a working parallel HDF5/h5py/mpi4py environment can be | ||
| challenging and is often specific to your local computing resources, | ||
| e.g., the supercomputer that you're running on typically already has | ||
| its preferred MPI installation. As a starting point we provide | ||
| instructions that worked in a specific, fairly generic environment. | ||
|
|
||
| These instructions successfully built parallel HDF5/h5py | ||
|
|
@@ -133,8 +133,8 @@ | |
| CC=mpicc HDF5_DIR=$HDF5_PATH python setup.py build | ||
| python setup.py install | ||
|
|
||
| If you have questions or want to share how you managed to build | ||
| parallel hdf5/h5py/mpi4py please let everyone know on the | ||
| `MDAnalysis forums`_. | ||
|
|
||
| .. _`H5MD`: https://nongnu.org/h5md/index.html | ||
|
|
@@ -180,6 +180,9 @@ | |
|
|
||
| .. automethod:: H5MDReader._reopen | ||
|
|
||
| .. autoclass:: H5PYPicklable | ||
| :members: | ||
|
|
||
| """ | ||
|
|
||
| import numpy as np | ||
|
|
@@ -190,6 +193,15 @@ | |
| import h5py | ||
| except ImportError: | ||
| HAS_H5PY = False | ||
|
|
||
| # Allow building documentation even if h5py is not installed | ||
| import types | ||
|
|
||
| class MockH5pyFile: | ||
| pass | ||
| h5py = types.ModuleType("h5py") | ||
| h5py.File = MockH5pyFile | ||
|
|
||
| else: | ||
| HAS_H5PY = True | ||
|
|
||
|
|
@@ -228,23 +240,23 @@ class H5MDReader(base.ReaderBase): | |
|
|
||
| See `h5md documentation <https://nongnu.org/h5md/h5md.html>`_ | ||
| for a detailed overview of the H5MD file format. | ||
|
|
||
| The reader attempts to convert units in the trajectory file to | ||
| the standard MDAnalysis units (:mod:`MDAnalysis.units`) if | ||
| `convert_units` is set to ``True``. | ||
|
|
||
| Additional data in the *observables* group of the H5MD file are | ||
| loaded into the :attr:`Timestep.data` dictionary. | ||
|
|
||
| Only 3D-periodic boxes or no periodicity are supported; for no | ||
| periodicity, :attr:`Timestep.dimensions` will return ``None``. | ||
|
|
||
| Although H5MD can store varying numbers of particles per time step | ||
| as produced by, e.g., GCMC simulations, MDAnalysis can currently | ||
| only process a fixed number of particles per step. If the number | ||
| of particles changes a :exc:`ValueError` is raised. | ||
|
|
||
| The :class:`H5MDReader` reads .h5md files with the following | ||
| HDF5 hierarchy: | ||
|
|
||
| .. code-block:: text | ||
|
|
@@ -277,21 +289,21 @@ class H5MDReader(base.ReaderBase): | |
| \-- (position) | ||
| \-- [step] <int>, gives frame | ||
| \-- [time] <float>, gives time | ||
| +-- unit <str> | ||
| \-- [value] <float>, gives numpy arrary of positions | ||
| with shape (n_atoms, 3) | ||
| +-- unit <str> | ||
| \-- (velocity) | ||
| \-- [step] <int>, gives frame | ||
| \-- [time] <float>, gives time | ||
| +-- unit <str> | ||
| \-- [value] <float>, gives numpy arrary of velocities | ||
| with shape (n_atoms, 3) | ||
| +-- unit <str> | ||
| \-- (force) | ||
| \-- [step] <int>, gives frame | ||
| \-- [time] <float>, gives time | ||
| +-- unit <str> | ||
| \-- [value] <float>, gives numpy arrary of forces | ||
| with shape (n_atoms, 3) | ||
| +-- unit <str> | ||
|
|
@@ -557,13 +569,15 @@ def open_trajectory(self): | |
| if self._comm is not None: | ||
| # can only pass comm argument to h5py.File if driver='mpio' | ||
| assert self._driver == 'mpio' | ||
| self._file = H5PYPicklable(name=self.filename, # pragma: no cover | ||
| mode='r', | ||
| driver=self._driver, | ||
| comm=self._comm) | ||
| elif self._driver is not None: | ||
| self._file = H5PYPicklable(name=self.filename, mode='r', | ||
| driver=self._driver) | ||
| else: | ||
| self._file = H5PYPicklable(name=self.filename, mode='r') | ||
| # pulls first key out of 'particles' | ||
| # allows for arbitrary name of group1 in 'particles' | ||
| self._particle_group = self._file['particles'][ | ||
|
|
@@ -727,3 +741,88 @@ def has_forces(self): | |
| @has_forces.setter | ||
| def has_forces(self, value: bool): | ||
| self._has['force'] = value | ||
|
|
||
| def __getstate__(self): | ||
| state = self.__dict__.copy() | ||
| del state['_particle_group'] | ||
| return state | ||
|
|
||
| def __setstate__(self, state): | ||
| self.__dict__ = state | ||
| self._particle_group = self._file['particles'][ | ||
| list(self._file['particles'])[0]] | ||
| self[self.ts.frame] | ||
|
|
||
|
|
||
| class H5PYPicklable(h5py.File): | ||
| """H5PY file object (read-only) that can be pickled. | ||
|
|
||
| This class provides a file-like object (as returned by | ||
| :class:`h5py.File`) that, | ||
| unlike standard Python file objects, | ||
| can be pickled. Only read mode is supported. | ||
|
|
||
| When the file is pickled, filename, mode, driver, and comm of | ||
| :class:`h5py.File` in the file are saved. On unpickling, the file | ||
| is opened by filename, mode, driver. This means that for a successful | ||
| unpickle, the original file still has to be accessible with its filename. | ||
|
|
||
| Parameters | ||
| ---------- | ||
| filename : str or file-like | ||
| a filename given a text or byte string. | ||
| driver : str (optional) | ||
| H5PY file driver used to open H5MD file | ||
|
|
||
| Example | ||
| ------- | ||
| :: | ||
|
|
||
| f = H5PYPicklable('filename', 'r') | ||
| print(f['particles/trajectory/position/value'][0]) | ||
| f.close() | ||
|
|
||
| can also be used as context manager:: | ||
|
|
||
| with H5PYPicklable('filename', 'r'): | ||
| print(f['particles/trajectory/position/value'][0]) | ||
|
|
||
| Note | ||
| ---- | ||
| Pickling of an `h5py.File` opened with `driver="mpio"` and an MPI | ||
| communicator is currently not supported | ||
|
|
||
| See Also | ||
| --------- | ||
| :class:`MDAnalysis.lib.picklable_file_io.FileIOPicklable` | ||
| :class:`MDAnalysis.lib.picklable_file_io.BufferIOPicklable` | ||
| :class:`MDAnalysis.lib.picklable_file_io.TextIOPicklable` | ||
| :class:`MDAnalysis.lib.picklable_file_io.GzipPicklable` | ||
| :class:`MDAnalysis.lib.picklable_file_io.BZ2Picklable` | ||
|
|
||
|
|
||
| .. versionadded:: 2.0.0 | ||
| """ | ||
|
|
||
| def __getstate__(self): | ||
| driver = self.driver | ||
| # Current issues: Need a way to retrieve MPI communicator object | ||
| # from self and pickle MPI.Comm object. Parallel driver is excluded | ||
| # from test because h5py calls for an MPI configuration when driver is | ||
| # 'mpio', so this will need to be patched in the test function. | ||
| if driver == 'mpio': # pragma: no cover | ||
| raise TypeError("Parallel pickling of `h5py.File` with" # pragma: no cover | ||
| " 'mpio' driver is currently not supported.") | ||
|
|
||
| return {'name': self.filename, | ||
|
Member
There was a problem hiding this comment. I am confused: If the
Member
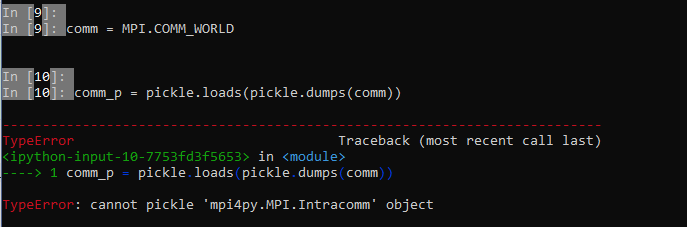
There was a problem hiding this comment. I see that you had it included in b29e675 but then removed again. Did you test that a from mpi4py import MPI
import pickle
comm = MPI.COMM_WORLD
comm_p = pickle.loads(pickle.dumps(comm))
assert comm == comm_por something similar?
Member
Author
There was a problem hiding this comment. That does not seem to work
Contributor
There was a problem hiding this comment. I don’t have a working mpi4py in my desktop. But it seems that it cannot be pickled natively. |
||
| 'mode': self.mode, | ||
| 'driver': driver} | ||
|
|
||
| def __setstate__(self, state): | ||
| self.__init__(name=state['name'], | ||
| mode=state['mode'], | ||
| driver=state['driver']) | ||
|
|
||
| def __getnewargs__(self): | ||
| """Override the h5py getnewargs to skip its error message""" | ||
| return () | ||
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains hidden or bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Add this suggestion to a batch that can be applied as a single commit.
This suggestion is invalid because no changes were made to the code.
Suggestions cannot be applied while the pull request is closed.
Suggestions cannot be applied while viewing a subset of changes.
Only one suggestion per line can be applied in a batch.
Add this suggestion to a batch that can be applied as a single commit.
Applying suggestions on deleted lines is not supported.
You must change the existing code in this line in order to create a valid suggestion.
Outdated suggestions cannot be applied.
This suggestion has been applied or marked resolved.
Suggestions cannot be applied from pending reviews.
Suggestions cannot be applied on multi-line comments.
Suggestions cannot be applied while the pull request is queued to merge.
Suggestion cannot be applied right now. Please check back later.
Uh oh!
There was an error while loading. Please reload this page.